
Abstract
In some wild Atlantic salmon populations, rapid declines in numbers of wild returning adults has been associated with an increase in the prevalence of farmed salmon. Studies of phenotypic variation have shown that interbreeding between farmed and wild salmon may lead to loss of local adaptation. Yet, few studies have attempted to assess the impact of interbreeding at the genome level, especially among North American populations. Here, we document temporal changes in the genetic makeup of the severely threatened Magaguadavic River salmon population (Bay of Fundy, Canada), a population that might have been impacted by interbreeding with farmed salmon for nearly 20 years. Wild and farmed individuals caught entering the river from 1980 to 2005 were genotyped at 112 single-nucleotide polymorphisms (SNPs), and/or eight microsatellite loci, to scan for potential shifts in adaptive genetic variation. No significant temporal change in microsatellite-based estimates of allele richness or gene diversity was detected in the wild population, despite its precipitous decline in numbers over the last two decades. This might reflect the effect of introgression from farmed salmon, which was corroborated by temporal change in linkage-disequilibrium. Moreover, SNP genome scans identified a temporal decrease in candidate loci potentially under directional selection. Of particular interest was a SNP previously shown to be strongly associated with an important quantitative trait locus for parr mark number, which retained its genetic distinctiveness between farmed and wild fish longer than other outliers. Overall, these results indicate that farmed escapees have introgressed with wild Magaguadavic salmon resulting in significant alteration of the genetic integrity of the native population, including possible loss of adaptation to wild conditions.
Similar content being viewed by others
Introduction
Farmed fish are reared in environments where selective pressures are directed toward improving commercially important traits, namely rapid growth and older age at maturity (Theodorou and Couvet, 2004). Consequently, farmed salmon are known to exhibit pronounced differences with wild counterparts at many phenotypic traits, such as growth rate, aggressiveness, predator avoidance behavior, acid tolerance (Jonsson and Jonsson 2006; Fraser et al., 2008), which are likely associated with reduced reproductive success in the wild. Moreover, relaxed selection resulting from absence of predators, constant supply of food and medical treatment, as well as increased genetic drift caused by small effective population sizes may favor the accumulation of deleterious alleles. Furthermore, genetic changes unrelated to adaptation to captivity are bound to occur as a result of founder effects in the farmed populations (Roberge et al., 2006). Consequently, change in the genetic composition of captive populations has been observed in many studies on farmed Atlantic salmon of northern Europe (Skaala et al., 2004). Nevertheless, farmed salmon reproduce and hybridize with wild conspecifics, which raises major concerns for conservation programs aimed at protecting the genetic integrity of already diminished populations (Hutchings and Fraser, 2008).
Indeed, the impacts of farmed fish escaping and mixing with wild populations represent a serious threat to natural populations (Fleming et al., 2000; McGinnity et al., 2003; Theodorou and Couvet, 2004). Potential alteration of the genetic composition of the wild population impacted by escapees may have significant consequences in terms of reduction of potential to respond to changing environments. Moreover, studies of phenotypic variation have shown that interbreeding between farmed and wild salmon may lead to a reduction in fitness and local adaptation, ultimately contributing to the possible extinction of wild populations (McGinnity et al., 2003; Fraser et al., 2008). Introgressive hybridization between farm and wild salmon also lead to misregulation of gene expression in the latter (Roberge et al., 2008; Normandeau et al., 2009). Yet, despite the potential genetic consequences arising from the introduction of farmed salmon, few studies have attempted to assess the impact of gene flow from farmed Atlantic salmon to wild Atlantic salmon populations at the genetic level. Such studies require long-term monitoring of impacted populations, and are rare, especially in North America.
Many wild Atlantic salmon (Salmo salar) populations of the Bay of Fundy region (Canada) have collapsed since the 1980s and are now threatened with extirpation or have already been extirpated (DFO, 2009). Causes for this rapid decline may include changes in oceanographic conditions, freshwater habitat degradation as well as impacts from the Bay of Fundy's commercial Atlantic salmon aquaculture industry (Carr et al., 2004). The Magaguadavic River, located in the Bay of Fundy region (Canada) has been monitored by local authorities since the establishment of the aquaculture industry over 30 years ago. The production of farmed salmon increased dramatically over the last 20 years, resulting in large numbers of fish escaping into the wild. Indeed, in the mid-1990s, farmed salmon entering the Magaguadavic River far exceeded the number of wild salmon (Carr et al., 1997). Farmed females have also been shown to successfully spawn in the river. In 1993 alone, up to 55% of the redds were at least partially of farmed origin, despite a greater number of wild salmon entering the river compared with farmed salmon (Carr et al., 1997). At present, wild salmon represent a small proportion of salmon entering the river, whereas farmed fish are still abundant in the system. Clearly, the Magaguadavic River wild Atlantic salmon population may be facing two major challenges in regard to protecting its genetic integrity; a declining census population size and possible interbreeding with farmed salmon.
The main goal of this study was to document temporal change in the genetic composition of the Magaguadavic River wild Atlantic salmon population following the rapid increase in the number of farmed salmon entering the river from 1980 to the early 2000s. We used both microsatellite and single-nucleotide polymorphism (SNP) markers (comprising both putatively adaptive and neutral markers) to detect the possible effects of interbreeding between wild and farmed salmon at the genetic level. Under the assumption of admixture between the two groups, we first expected neutral markers to exhibit a lower diversity in the domesticated strain and consequently reduced diversity over time in wild introgressed individuals. As the domestic strain originated from the nearby Saint John River (approximately 60 km from the Magaguadavic River), we also expected the genetic markers to naturally reveal weak divergence at the majority of markers between farm and wild fish. However, we expected strong divergence at some markers as a result of the radically different selective pressures in captive and wild environments indicated by markers potentially affected (either directly or indirectly through linkage) by divergent or directional selection. Finally, since evidence of farmed salmon spawning in this system has been shown earlier, if farm salmon have indeed been interbreeding with wild salmon through the 1990s, we expected genetic divergence between these two groups of salmon to be reduced over time as a result of introgression.
Materials and methods
Sample collection
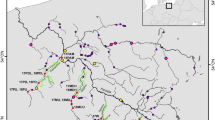
Samples were collected from wild and farmed salmon captured in the Magaguadavic River between 1980 and 2005. Wild adults were captured at the head of tide fishway trap after ascending the river (Figure 1). From mid-April to June 2000 smolts were either trapped as they descended the river using a fykenet in a small stream serving as the downstream fish bypass outlet for the head of tide hydroelectric dam, or they were collected 24 km upstream of the dam using a rotary screw trap (for example, Solutions Inc., Corvallis, OR, USA). Adult salmon used in this study were identified as wild, except in 1992 and 2000 when both wild and farmed individuals were sampled. Wild and farmed smolts were sampled in 2000. Adults and smolts were classified as wild or farmed origin using morphological features such as fin and gill cover erosion (associated with the farming environment), and by examining circuli patterns on scales as previously described by Carr et al. (1997) and Carr and Whoriskey (2006). Blood was collected and frozen for samples from 1992 and either air-dried scale and/or fin clips conserved in 95% ethanol were collected from individuals captured in 1980, and from 1996 to 2005. In summary, the sample collections (also called populations hereafter) used were the following: (1) WILD-1980, (2) WILD-1992, (3) FARMED-1992, (4) WILD-1996, (5) WILD-1998_99, (6) WILD-2000_adults, (7) WILD-2000_smolts, (8) FARMED-2000_adults, (9) FARMED-2000_smolts and (10) WILD-2002+.

Map showing the Magaguadavic River mouth and the fishway located close to the Passamaquoddy Bay where most of New Brunswick commercial salmon sea-cage sites are in the Bay of Fundy. Modified with permission from Carr et al. (1997) and Carr et al. (2004). (NB, New Brunswick; NS, Nova Scotia; PEI, Prince Edward Island; MA, Maine).
DNA genotyping
The microsatellite and SNP analyses carried out here involved two somewhat overlapping but different groups of samples, primarily because of the unsuitability of older samples for SNP analyses. Organization of samples for analyses is summarized in Table 1. A group composed exclusively of wild adults caught in 1980 (WILD-1980) (n=31), 1992 (WILD-1992) (n=60) and 2000 (WILD-2000_adults) (n=34), farmed adults from 2000 (FARMED-2000_adults) (n=53) as well as wild and farmed smolts sampled in 2000 (WILD-2000_smolts, FARMED-2000_smolts) (n=48 and n=39, respectively) was genotyped for microsatellites only (referred to as group A). A second group was genotyped for SNPs (referred to as group B). This group was organized into different clusters based on aquaculture history in the Bay of Fundy as follows: farm escapees from early-aquaculture 1992 (FARMED-1992) (n=20), wild adults from the early-aquaculture period in 1992 (WILD-1992) (n=18), wild adults from the mid-aquaculture period in 1995–1996 (WILD-1996) (n=20), wild adults from the late-aquaculture period in 1998–1999 (WILD-1998_99) (n=22) and wild adults from the very late-aquaculture period in 2002+ (WILD-2002+) (n=15). Note that all individuals analyzed for SNP variation were also genotyped at the same microsatellite loci as were the samples in group A and the group B WILD-1992 sample collection was a subset of the samples contained in the WILD-1992 collection.
DNA was extracted using Qiagen 96-well DNeasy plates, following the manufacturer's specifications (Qiagen, Valencia, CA, USA). We genotyped individuals at eight microsatellite loci. PCR amplifications were carried out in 10 μl volumes, containing between 1 and 100 ng of template DNA, 2 mM each dNTP, 0.5 μM labelled and unlabelled primers, 50 mM KCl, 0.5 units of Taq DNA polymerase supplied by MBI Fermentas (Burlington, ON, Canada) and 2.0 mM MgCl2. Thermal cycling conditions were as follows: (94 °C for 3 min) × 1, (94 °C for 1 min, 58 °C for 30 s and 72 °C for 30 s) × 5 and (90 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s) × 30, followed by a 15-min extension step at 72 °C. Primer sequences for loci Ssa 197 and Ssa 202 are given in O'Reilly et al. (1996), SSsp 2201, SSsp 2210, SSsp 2215, SSsp 2216, SSsp 1G7 and SSsp 1605 are given in Paterson et al. (2004). PCR products were combined, and salt, unincorporated dNTPs, unincorporated labelled and non-labelled primers were removed using Qiagen's 96 MinElute 96 UF PCR purification kits, following the manufacturer's procedures. Fragments were size fractionated and detected using an Applied Biosystems 3130 XL (Applied Biosystems, Carlsbad, CA, USA). One sample from each strip of eight tubes was duplicated to identify sample placement errors, strip inversions and plate inversions. In all, 2 of 10 laboratory samples were analyzed in each group of 96 samples both to identify individual batches, and to ensure allele calls were standardized across batches of samples analyzed over time.
SNP genotyping was done using the iPlex Gold assay on the MassARRAY platform (Sequenom, San Diego, CA) according to the manufacturer's instructions. In total, a panel of 388 Atlantic salmon SNPs were genotyped in 16 different multiplexes, with multiplexing levels ranging from 12 to 36. The 13 multiplexes with the highest number of SNPs from Moen et al. (2008) as well as the 3 multiplexes with the highest number of SNPs from Lorenz et al. (2010) were selected for genotyping. PCR and extension primers were designed with the software MassARRAY AssayDesign v3.1 (Sequenom). Allele separations were performed using the Sequenom MassARRAY Analyzer. Genotypes were assigned in real time on the basis of the mass peaks (Tang et al., 1999) using the MassARRAY SpectroTYPER RT v3.4 software (Sequenom). Manual inspection of all the results was carried out using the MassARRAY TyperAnalyzer v4.0 software (Sequenom). Individuals were genotyped only once for each multiplex since previous experiences with the SNPs revealed no discrepancies between replicate analyses (CIGENE, unpublished information). Moreover, the genotyping was followed by highly stringent quality control criteria to keep only high quality markers for further analyses (see results).
Genetic variation and differentiation
For comparative purposes, we considered separate temporal replicates as individual populations in subsequent analyses. For microsatellite analysis, populations of group B were considered with group A with the exception of WILD-1992. Neutral microsatellite genetic variation within each population was quantified using standard descriptive statistics for each locus individually and globally: observed and expected heterozygosities (HO and HE) as well as Weir and Cockerham (1984) FIS values were estimated using GENETIX (Belkhir et al., 2001), and the number of observed alleles as well as Nei's (1977) estimator of gene diversity, using FSTAT 2.9.3 (Goudet et al., 2002). We calculated allelic richness (Â), which is the number of different alleles corrected for variation in sample size, using the rarefaction method employed by FSTAT 2.9.3 to the smallest sample (n=11). Locus SSsp 2201 was excluded from the calculation of allelic richness to increase the power of detecting differences in  (Leberg, 2002) between populations because the sample size would have become too small had it included locus SSsp 2201 (n=3). The distributions of within population gene diversity did not all conform to normality after Shapiro–Wilk tests (four tests with P<0.01). On the other hand, allelic richness distribution all conformed to normality after Shapiro–Wilk tests (all P>>0.01) and homogeneity of variance among populations was verified using a Bartlett test (K2=0.759, df=9, P=0.999). Therefore, respectively, a Kruskal–Wallis analysis of variance (non-parametric) and an analysis of variance (parametric) were used to determine if gene diversity and allelic richness changed among samples. Conformity of individual loci to Hardy–Weinberg equilibrium (HWE) expectations was also tested using FSTAT 2.9.3 and Fisher's combined probabilities across loci for each population was used to determine a global P-value for the set of HWE tests (Mosteller and Fisher, 1948). Genic differentiation (G; Guo and Thompson, 1992) at individual loci between all pairs of populations and significance values over all loci were obtained using Fisher's method (Ryman and Jorde, 2001) as implemented using GENEPOP 3.4 (Raymond and Rousset, 1995). The sequential Bonferroni correction for multiple tests was applied to maintain the table-wide significance level at α=0.05 (Rice, 1989) while testing for HWE and genic differentiation. After removal of putatively non-neutral loci identified in FDIST2 analyses (Beaumont and Nichols, 1996) (see below), microsatellites and SNP markers were used separately for calculations of pair-wise genetic differentiation between populations using the FST of Weir and Cockerham (1984) (θ) as calculated with GENETIX after 1000 permutations for significance.
Linkage disequilibrium
We do not present any admixture analysis, for instance based on clustering methods (for example, Structure, Pritchard et al., 2000). Although such attempts were made (results not shown), the low level of differentiation observed for the vast majority of markers used (see Results section) resulted in insufficient power to apply such methods adequately (Vaha and Primmer, 2006). On the other hand, as natural outbred populations are expected to be in near genome-wide linkage equilibrium relative to a situation of interbreeding with an invasive population, which is expected to generate linkage disequilibrium (LD) in a recently introgressed population (Gaut and Long, 2003; Allendorf and Luikart, 2007), we used ARLEQUIN 3.5 (Excoffier and Lischer, 2010) to test for pair-wise LD within populations genotyped for SNPs. More specifically, we tested for the presence of significant associations between pairs of loci, based on a likelihood ratio test, wherein the likelihood of observing the sample evaluated under the hypothesis of no association between loci (linkage equilibrium) is compared with the likelihood of observing the sample when association is allowed (Excoffier and Lischer, 2010). When the likelihood of an association between two loci is significantly greater than no association, they are considered to be linked or under LD. The distributions of the numbers of linked loci within populations deviated significantly from normality (Shapiro–Wilk tests, all P<0.01). Therefore, the median number of loci under LD per locus within each population was compared among populations with a Kruskal–Wallis analysis of variance to determine if LD changed across temporal samples of wild Magaguadavic Atlantic salmon. When significant differences among groups were observed, Wilcoxon signed-rank tests were used to compare median values between populations and determine which samples differed. Furthermore, we used MULTILOCUS (Agapow and Burt, 2001) to calculate rd, the multilocus LD within population, as a measure of genome-wide LD over all SNP markers.
Genome scans
Numerous methods to detect loci potentially under the effects of natural selection have been developed over the last decade, most based on principles reported by Lewontin and Krakauer (1973). Some of these methods proposed to compare levels of genetic diversity and differentiation between populations as loci under directional selection should show larger differences, and loci under balancing selection should present less divergence between populations than neutrally evolving loci. FDIST, (Beaumont and Nichols, 1996), was one of the first programs widely available to test for departures from neutrality, and the analyses carried out have become widely known as ‘genome scans’. In the latest version of FDIST2, simulations under a finite island-model are performed to obtain a null distribution of FST values across loci as a function of heterozygosity, and loci with an unusually high or low FST value (‘outliers’) are generally considered to be potentially under the effect of natural selection (either directly or indirectly through linkage). Here, we scanned genetic variation at SNP markers by comparing aquaculture samples to all wild temporal samples to detect changes in loci potentially under divergent selection. We used a confidence interval of 0.95 for the expected null differentiation meaning that loci over this interval had to be in the upper 0.025 tail of the distribution to be considered as potentially under directional or divergent selection or in the lower 0.025 tail of the distribution to be considered as potentially under balancing selection.
Results
Genetic variation
Over all samples, all microsatellite loci were highly polymorphic, with the total number of alleles per locus ranging from 11 to 36 (mean=24.38), and observed heterozygosity per locus across populations ranging from 0.5 to 1.0 (mean=0.831) (Supplementary Table S1). Mean gene diversity per locus per population ranged from 0.831 to 0.878 and allelic richness from 1 to 20 alleles per locus per population. Neither gene diversity nor allelic richness were significantly different among samples (Kruskal–Wallis K2=2.759, df=9 and P=0.973; analysis of variance F=0.019, df=1 and P=0.891, respectively). The null hypothesis of HWE was not rejected for any locus and for any population after correcting for multiple tests (α=0.000625, k=80). Fisher's combined probabilities across loci was not significant for seven out of eight populations (P>0.05) with only FARM-2000_adults presenting significant departure from HWE (P=0.001) and with small but positive FIS indicative of a slight heterozygote deficiency (Supplementary Table S1).
Out of the 388 SNP (Supplementary Table S2) assayed, 348 markers yielded at least one genotype but only 267 markers were polymorphic. Consequently, 81 were discarded for further analyses. Markers with call rate inferior to 95% (54) and minor allele frequencies <0.05 (101) were also excluded to ensure only high-quality informative markers were utilized. Thus, the final group of SNPs used in all subsequent analyses numbered 112, and average expected and observed heterozygosities are given in Table 1.
Differentiation
Pair-wise genic tests of population differentiation at individual microsatellite loci yielded only 26 significant comparisons out of 360 after correction for multiple comparisons (α=0.000139, k=360). This translated into a nonsignificant (P=0.999) global FST value of 0.00096, reflecting an overall very weak level of differentiation among populations. Furthermore, microsatellite pair-wise FST values (θ) were significant in only 20 out of 45 comparisons (Table 2) and ranged from 0.005 between WILD-2000_smolts and FARM-2000_adults to 0.019 between WILD-1980 and FARM-2000_adults and between WILD-1996 and WILD-2002+ when significant. The WILD-1980 population was associated with the greatest number of significant pair-wise FST estimates (eight of nine), and the largest FST values, ranging from 0.009 to 0.018 when compared with other wild population temporal replicates, and from 0.011 to 0.019 when compared with aquaculture sample collections. The sample collection WILD-2002+ was associated with the next largest FST values, ranging from 0.010 to 0.019 when compared against other wild temporal replicates, of which four of five were significant (WILD-1992, WILD-1996, WILD-2000_adults and WILD-2000_smolts). The lowest significant FST values based on microsatellite data were observed for pair-wise comparisons between wild replicates post-1980 and aquaculture samples (range between 0.005 and 0.009). For SNP markers, pair-wise FST values varied between 0.000 (in 5/10 comparisons) and 0.006 (between WILD-1996 and FARM-1992) (results not shown). None of these pair-wise FST values were significant after removal of six SNPs identified by FDIST2 as potentially under divergent selection in a global test over all samples at a significant level of 0.025 (results not shown).
Linkage disequilibrium
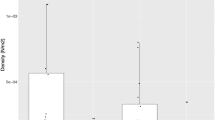
SNP markers yielded variable patterns of LD in wild temporal replicates and the 1992 farm salmon samples (Figure 2). The Kruskal–Wallis test of equality of medians among populations was significant (K2=139.470, df=4 and P<<0.001). Pair-wise comparisons showed that FARM-1992 and WILD-1996 as well as WILD-1998_99 and WILD-2002+ exhibited similar numbers of linked loci per locus (W=6686 and 6753 with P=0.394 and 0.321, respectively) while all eight other comparisons were significantly different after corrections for multiple comparisons (P<<0.001). Multilocus LD (rd) values within each population are given in Figure 2 and corroborate the detailed locus-by-locus LD analyzes within populations in the sense that all post-1992 wild temporal samples had higher rd values than the 1992 wild sample, and the aquaculture sample (FARM-1992) presented the highest multilocus LD.

Number of SNP-linked loci per individual SNP locus within population for: (a) FARM-1992, (b) WILD-1992, (c) WILD-1996 (d) WILD -1998_99 and (e) WILD-2002+. Loci are not arranged in the same order among populations but by decreasing order according to the x axis. Median number of linked loci per locus per population (M) and multilocus rd per population are indicated on each panel. Within population M are not different for populations sharing the same capital letter in the bottom-right corner of panels after Wilcoxon tests (see Results section).
FDIST outlier detection test
Genome scan plots of marker FST as a function of heterozygosity comparing each of the four wild samples with the farmed salmon (FARM-1992) are shown in Figure 3, with outliers potentially under divergent selection over the 95% confidence level. Single locus FST values for pair-wise comparisons were approximately 0.01, varying slightly from locus to locus (between 0.006 for WILD-1998_99 and 0.011 for WILD-1996) whereas outlier FST values were all above 0.10. Overall, we found a twofold reduction in the number of observed markers potentially under the effect of divergent selection after 1996, with the observed number of outliers, in chronological sequence, of 10 (WILD-1992), 9 (WILD-1996), 4 (WILD-1998_99) and 5 (WILD-2002+). Table 3 presents a summary of these markers, including their names, known linkage group used from Boulding et al. (2008) and Lorenz et al. (2010), as well as gene annotation when available. Three outliers (Contig14899_0107, Contig15610_550 and Contig16686_0431) identified in the genome scan involving WILD-1992, were also identified in comparisons with WILD-1995_96 and/or WILD-1998_99. Of these markers, Contig14899_0107 presented a nearly significant reduction in FST through time (r2=0.88, P=0.062), and was no longer an outlier in the WILD-2002+ genome scan (Figure 4). Loci showing less differentiation than that represented by the lower 95% confidence interval would normally be considered as potentially under balancing selection, but as this confidence level always fell below the FST=0, we rejected the hypothesis of these markers being under the effect of balancing selection.

Differentiation (FST) as a function of heterozygosity as calculated by FDIST2 when comparing FARM-1992 with: (a) WILD-1992, (b) WILD-1996 (c) WILD -1998_99 and (d) WILD-2002+. On each panel, solid line represent upper and lower 95% confidence level and dotted line indicates the average FST across loci. Asterisk (*) on outliers indicate two values represented by the same dot on the graphic (double asterisks (**) three values). Only SNP markers were used.

Differentiation (FST) of Contig14899_0107 across time as estimated by FDIST2 in genome scans (Figure 3). Wild temporal samples were considered as single year sample based on the sample year predominant in the population (WILD-1996=1996, WILD -1998_99=1999 and WILD-2002+=2002). Regression value is indicated in the top-right corner (P=0.062).
Discussion
This study is the first to examine the genetic impacts of introgression from farmed Atlantic salmon into a wild population using a wide panel of SNP, in addition to known neutral microsatellite markers. The long-term monitoring of the Magaguadavic River of the Bay of Fundy in Eastern Canada permitted a comparison of the genetic composition of wild samples before and after the establishment and growth of the salmon farming industry in the area. More specifically, we assessed whether introgression affected (i) levels of neutral genetic diversity in wild Magaguadavic River Atlantic Salmon over time, (ii) the extent of differentiation between wild and farmed replicates at both types of markers, (iii) the signature of divergent selection possibly operating at some SNP loci and (iv) the initial degree of distinctiveness or conversely, the homogenization of wild and farmed salmon. Our results strongly suggest that introgression did change the genetic signature of the wild population, by, for example, reducing the divergence at neutral and non-neutral markers. Although levels of within population genetic diversity in the wild population did not appear to change, levels of differentiation between wild and farmed salmon were reduced through time, likely in response to the increasing presence and influence of farmed salmon in the Magaguadavic River. Perhaps more importantly, the overall number of loci potentially under divergent selection was reduced when wild temporal samples were contrasted with a reference farmed sample. Furthermore, a specific marker putatively involved in local adaptation (see Discussion below) epitomized the homogenizing effect of introgression by showing a reduction of differentiation in wild populations compared with farmed samples over time, until patterns of locus variability and population differentiation at this locus were indistinguishable from that expected because of neutral evolutionary forces.
Neutral genetic changes
The unexpected stability of levels of gene diversity and allelic richness of wild temporal replicates might have been foreseen in the light of introgression by a similarly diverse group of individuals and consistent input from this group. Previous studies of European domesticated fish strains showed reduced allelic variation compared with their European wild relatives (Mjølnerød et al., 1997; Norris et al., 1999; Skaala et al., 2004). Furthermore, as farmed salmon typically involve a restricted number of effective breeder, selection for performance traits and low or no gene flow, we expected farmed Atlantic salmon to show this kind of reduced genetic variation. However, we found that farmed salmon caught entering or descending the river as smolts did not exhibit lower levels of allelic richness or gene diversity compared with wild salmon. Although this remains to be rigorously tested, it is possible that some of the numerous freshwater aquaculture facilities each has lost and retained different alleles over time, and that collectively, they contain a large standing pool of genetic variation. These stocks would have acted partly as a metapopulation in neutralizing any possible effect of an isolated group of founders or of genetic drift.
Admixture from farm escapees in the wild may threaten natural populations through ‘maladaptive’ genetic changes (Skaala et al., 2006). Starting between 1992 and 1996, there were indications that introgression occurred in the wild Magaguadavic population. This was illustrated by the number of linked loci per locus in the wild replicate sample collections that went from a relatively stable state corresponding to genome-wide equilibrium to numerous LD strongly suggestive of a blending between two somewhat different populations. Indeed, the number of linked loci per locus in the wild sample from 1996 was similar to that observed in the farmed sample, which was possibly attributable to some variability in the origins or allele frequency distributions among the farmed population within the Magaguadavic River system. Moreover, this time period corresponded with the expansion of the farming industry in the Magaguadavic River and the observed turning point in relative abundance of wild versus farmed Atlantic salmon returning to the river as adults (Carr et al., 2004). Population bottlenecks also have the potential to increase LD for a short period of time (Gaut and Long, 2003) but we would argue that in this study, although the rapid decline of the wild population might have contributed to the increase LD and cannot be ruled out, the sudden genome-wide increase in LD is more consistent with a scenario involving recombination imposed by admixture. Consequently, while after 1994 the number of farmed fish returning to the river outnumbered the rapidly declining wild Atlantic salmon population, our results suggest that the reduction of gene diversity expected in this declining population was counteracted by the outbreeding effect of introgression from a slightly divergent and genetically variable population of aquaculture salmon.
To our knowledge, the most comparable study to ours involving wild Atlantic salmon was carried out by Skaala et al. (2006) and showed a reduction of differentiation and genetic distances among wild populations affected by farm escapees for 20–30 years despite no genetic diversity change in wild populations. Using microsatellites, we found a modest albeit significant genetic difference between pre-aquaculture wild individuals from 1980 and all other wild replicates except for the very late-aquaculture period samples. Moreover, comparisons of wild temporal samples also yielded significant levels of differentiation. Of the five of seven significant comparisons involving pre- or early-aquaculture period samples and farmed fish, the highest value was reached between the oldest wild samples (WILD-1980 and WILD-1992) and most recent aquaculture adults (FARM-2000_adults). These results, along with the finding that the majority of nonsignificant genetic differences observed among the most recent (mid-aquaculture period and later) wild samples and aquaculture samples are consistent with a homogenization effect occurring between the farmed and wild population of the Magaguadavic River because of introgression. In similar studies, Koskinen et al. (2002) and Hansen et al. (2010) reached similar conclusions when they contrasted historical and contemporary replicates of, respectively, wild grayling and wild brown trout populations admixed with hatchery fishes; both observed a significant reduction in levels of differentiation between wild and hatchery strains reflecting recent admixture. In a recent study on brook charr (Salvelinus fontinalis), Marie et al. (2010) also documented a homogenization of populations structure among wild populations following stocking events with domestic fish, accompanied with a greater genetic similarity between wild and domestic fish. Our study yielded similar findings that admixture did reduce genetic differentiation between the wild and farm salmon despite no overall effect detected on the level of genetic diversity in the wild population.
Possible effects on local adaptation
Although the genome scans performed here can be criticized for the lack of fit to reality with respect to the underlying models leading to false positives (for example, inclusion of populations that have experienced recent and marked population bottlenecks (Foll and Gaggiotti, 2008), we nevertheless consider the use of this approach as useful in at least obtaining a qualitative measure of the number of loci highly divergent between farmed salmon and wild temporal replicates as a good indication of the proportions of genomic regions potentially under the effect of divergent selection in each comparison. Here, outliers were approximately one order of magnitude more differentiated than the average SNP marker analyzed in the two sample groups compared. Moreover, our results suggest a 50% reduction in the number of loci potentially under divergent selection between wild and farmed salmon after only 3–5 generations of significant admixture occurring in the wild population. Also, the locus with the most pronounced differentiation (FST=0.18) in the comparison between farmed and wild salmon in 1992 (early aquaculture period) was still identified as an outlier when compared in the mid- and late-aquaculture periods with a reduction of its differentiation value until it faded away into the neutral distribution in the most recent comparison. Admittedly, the finding of a single locus, Contig14899_0107, exhibiting this pattern over four time points is not unlikely to occur by chance, even given the high regression coefficient observed. However, this marker was associated with a quantitative trait locus for parr mark number in a previous study (Boulding et al., 2008). Parr marks have been suggested to be a variable trait important for predator avoidance in the wild, allowing juvenile Atlantic salmon to blend in with the local substrate of the river (for example, Donnelly and Dill, 1984). In summary, the large decrease in FST over four time points, the association of the locus with a previously identified quantitative trait locus, and the fact that it presented the highest differentiation in the original genome scan represents compelling evidence that the trend observed is biologically meaningful and did not occur purely by chance. Therefore, this particular case of homogenization between wild and aquaculture fish in addition to the previously demonstrated reduction for markers potentially under divergent selection, suggests that the wild population of Atlantic salmon in the Magaguadavic River likely suffer from a loss of local adaptation exacerbated by introgression from farmed salmon.
On the other hand, this reduced number of outliers might also reflect the fading of selected traits in farmed Atlantic salmon as there is no indication of the directionality for the divergent selection pressure underlying the high differentiation of these markers. However, why selection (both intentional and unintentional) in a captive environment would have caused an increase in genetic similarity with a given wild population is unclear. In any case, it could still be argued that regardless of whether the selected trait(s) represent both local adaptations to native river conditions in the wild population and the hatchery environment in the captive population, the homogenization most probably resulted in reduced adaptation to both environments. The admixture between both groups probably resulted in a breakdown of LD around divergently selected regions of the genome (Via and West, 2008) and incidentally caused the observed reduction of detected outliers regardless to the actual selection occurring in the wild, which would have to be very strong to counteract the recombination occurring following introgression. Of course, further studies (such as that carried out by Fraser et al., 2008), comparing fitness of wild and farmed–wild hybrid salmon, should be undertaken to measure fitness-related traits of differently introgressed individuals in association with their actual allelic composition. However, in an obligate non-invasive field study such as this one, we argue that until more important genomic resources are available, or another significant decrease in the cost of whole genome sequencing is realized, our results make a strong case for reduced local adaptation in a wild population impacted by farm escapees.
Perspectives for aquaculture management
Hutchings and Fraser (2008) recently debated the pros and cons of using aquaculture strains derived from local versus non-local wild populations for salmon farming for a given region. Adding to this debate, our study suggests that although we observed introgression from farm escapees in the wild population with significant loss of neutral differentiation and a reduction of the potential number of loci under divergent selection, neutral genetic variability still prevailed in a rapidly declining population. As there is evidence of captive-reared salmon establishing themselves outside of their native ranges (Volpe et al., 2000; Soto et al., 2001), despite the inferior fitness of farmed Atlantic salmon in natural environments (McGinnity et al., 2003), one could argue that a foreign strain might have had stronger impacts on the wild salmon in terms of changing the genetic composition of such a vulnerable population, impacting both neutral and selective divergence. Nevertheless, given the geographic and likely phylogenetic proximity of the wild and putative source population of the aquaculture strain analyzed here, detecting outlier loci was already evidence of differential selective pressures acting on farmed and wild salmon. In addition, their fading through time, especially when associated with an important quantitative trait locus for juvenile Atlantic salmon, certainly should be regarded as a warning for the potential loss of local adaptation in any kind of farming industries unintentionally releasing individuals in the natural environment
In conclusion, despite its relatively limited genomic coverage, this study emphasizes the potential importance of long-term monitoring of wild populations impacted by farmed escapees. Furthermore, in this context and with increasingly important genomic resources soon to become available for non-model species such as Atlantic salmon, more in depth studies could further disentangle the relative impacts of farmed escapees on the neutral and adaptive diversity of affected populations on a genome-wide basis.
References
Agapow PM, Burt A (2001). Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1: 101–102.
Allendorf FW, Luikart G (2007). Conservation and the Genetics of Populations. Blackwell Publishing: Oxford, UK.
Beaumont MA, Nichols RA (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B Biol Sci 263: 1619–1626.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2001). GENETIX 4.02, logiciel sous windows TM pour la génétique des populations.. Laboratoire Génome, Population, Interactions: CNRS UMR 5000. Université de Montpellier II: Montpellier, France.
Boulding EG, Culling M, Glebe B, Berg PR, Lien S, Moen T (2008). Conservation genomics of Atlantic salmon: SNPs associated with QTLs for adaptive traits in parr from four trans-Atlantic backcrosses. Heredity 101: 381–391.
Carr JW, Anderson JM, Whoriskey FG, Dilworth T (1997). The occurrence and spawning of cultured Atlantic salmon (Salmo salar) in a Canadian river. ICES J Mar Sci 54: 1064–1073.
Carr JW, Whoriskey F, O'Reilly P (2004). Efficacy of releasing captive reared broodstock into an imperilled wild Atlantic salmon population as a recovery strategy. J Fish Biol 65: 38–54.
Carr JW, Whoriskey FG (2006). The escape of juvenile farmed Atlantic salmon from hatcheries into freshwater streams in New Brunswick, Canada. ICES J Mar Sci 63: 1263–1268.
DFO (2009). Canada's Policy for Conservation of Wild Atlantic Salmon. (ed. DFO Canada).
Donnelly WA, Dill LM (1984). Evidence for Crypsis in Coho Salmon, Oncorhynchus-Kisutch (Walbaum), Parr - substrate color preference and achromatic reflectance. J Fish Biol 25: 183–195.
Excoffier L, Lischer HEL (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Fleming IA, Hindar K, Mjolnerod IB, Jonsson B, Balstad T, Lamberg A (2000). Lifetime success and interactions of farm salmon invading a native population. Proc R Soc Lond B Biol Sci 267: 1517–1523.
Foll M, Gaggiotti O (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180: 977–993.
Fraser DJ, Cook AM, Eddington JD, Bentzen P, Hutchings JA (2008). Mixed evidence for reduced local adaptation in wild salmon resulting from interbreeding with escaped farmed salmon: complexities in hybrid fitness. Evol Appl 1: 501–512.
Gaut BS, Long AD (2003). The lowdown on linkage disequilibrium. Plant Cell 15: 1502–1506.
Goudet J, Perrin N, Waser P (2002). FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices. Institut d'Écologie, Université de Lausanne: Lausanne.
Guo SW, Thompson EA (1992). Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48: 361–372.
Hansen MM, Meier K, Mensberg KLD (2010). Identifying footprints of selection in stocked brown trout populations: a spatio-temporal approach. Mol Ecol 19: 1787–1800.
Hutchings JA, Fraser DJ (2008). The nature of fisheries- and farming-induced evolution. Mol Ecol 17: 294–313.
Jonsson B, Jonsson N (2006). Cultured Atlantic salmon in nature: a review of their ecology and interaction with wild fish. ICES J Mar Sci 63: 1162–1181.
Koskinen MT, Sundell P, Piironen J, Primmer CR (2002). Genetic assessment of spatiotemporal evolutionary relationships and stocking effects in grayling (Thymallus thymallus, Salmonidae). Ecol Lett 5: 193–205.
Leberg PL (2002). Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11: 2445–2449.
Lewontin RC, Krakauer J (1973). Distribution of gene frequency as a test of theory of selective neutrality of polymorphisms. Genetics 74: 175–195.
Lorenz S, Brenna-Hansen S, Moen T, Roseth A, Davidson WS, Omholt SW et al. (2010). BAC-based upgrading and physical integration of a genetic SNP map in Atlantic salmon. Anim Genet 41: 48–54.
Marie AD, Bernatchez L, Garant D (2010). Loss of genetic integrity correlates with stocking intensity in brook charr (Salvelinus fontinalis). Mol Ecol 19: 2025–2037.
McGinnity P, Prodohl P, Ferguson K, Hynes R, Maoil NO, Baker N et al. (2003). Fitness reduction and potential extinction of wild populations of Atlantic salmon, Salmo salar, as a result of interactions with escaped farm salmon. Proc R Soc Lond B Biol Sci 270: 2443–2450.
Mjølnerød IB, Refseth UH, Karlsen E, Balstad T, Jakobsen KS, Hindar K (1997). Genetic differences between two wild and one farmed population of Atlantic salmon (Salmo salar) revealed by three classes of genetic markers. Hereditas 127: 239–248.
Moen T, Hayes B, Baranski M, Berg PR, Kjøglum S, Koop BF et al. (2008). A linkage map of the Atlantic salmon (Salmo salar) based on EST-derived SNP markers. BMC Genomics 9: 223.
Mosteller F, Fisher RA (1948). Questions and answers. Am Stat 2: 30–31.
Nei M (1977). F-statistics and analysis of gene diversity in subdivided populations. Ann Hum Genet 41: 225–233.
Normandeau E, Hutchings JA, Fraser DJ, Bernatchez L (2009). Population-specific gene expression responses to hybridization between farm and wild Atlantic salmon. Evol Appl 2: 489–503.
Norris AT, Bradley DG, Cunningham EP (1999). Microsatellite genetic variation between and within farmed and wild Atlantic salmon (Salmo salar) populations. Aquaculture 180: 247–264.
O'Reilly PT, Hamilton LC, McConnell SK, Wright JM (1996). Rapid analysis of genetic variation in Atlantic salmon (Salmo salar) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Can J Fish Aquat Sci 53: 2292–2298.
Paterson S, Piertney SB, Knox D, Gilbey J, Verspoor E (2004). Characterization and PCR multiplexing of novel highly variable tetranucleotide Atlantic salmon (Salmo salar L.) microsatellites. Mol Ecol Notes 4: 160–162.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Raymond M, Rousset F (1995). An exact test for population differentiation. Evolution 49: 1280–1283.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Roberge C, Einum S, Guderley H, Bernatchez L (2006). Rapid parallel evolutionary changes of gene transcription profiles in farmed Atlantic salmon. Mol Ecol 15: 9–20.
Roberge C, Normandeau E, Einum S, Guderley H, Bernatchez L (2008). Genetic consequences of interbreeding between farmed and wild Atlantic salmon: insights from the transcriptome. Mol Ecol 17: 314–324.
Ryman N, Jorde PE (2001). Statistical power when testing for genetic differentiation. Mol Ecol 10: 2361–2373.
Skaala O, Hoyheim B, Glover K, Dahle G (2004). Microsatellite analysis in domesticated and wild Atlantic salmon (Salmo salar L.): allelic diversity and identification of individuals. Aquaculture 240: 131–143.
Skaala O, Wennevik V, Glover KA (2006). Evidence of temporal genetic change in wild Atlantic salmon, Salmo salar L., populations affected by farm escapees. ICES J Mar Sci 63: 1224–1233.
Soto D, Jara F, Moreno C (2001). Escaped salmon in the inner seas, southern Chile: facing ecological and social conflicts. Ecol Appl 11: 1750–1762.
Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Köster H (1999). Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci USA 96: 10016–10020.
Theodorou K, Couvet D (2004). Introduction of captive breeders to the wild: harmful or beneficial? Conserv Genet 5: 1–12.
Vaha JP, Primmer CR (2006). Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15: 63–72.
Via S, West J (2008). The genetic mosaic suggests a new role for hitchhiking in ecological speciation. Mol Ecol 17: 4334–4345.
Volpe JP, Taylor EB, Rimmer DW, Glickman BW (2000). Evidence of natural reproduction of aquaculture-escaped Atlantic salmon in a coastal British Columbia river. Conserv Biol 14: 899–903.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population-structure. Evolution 38: 1358–1370.
Acknowledgements
We are grateful to M Best and E Merrill for their assistance in the field; to L Hamilton, M Cassista-da Ros, and L de Mestral Bezanson (DFO) for DNA extractions and microsatellite genotyping analysis. The SNP genotyping was performed by CIGENE at the national technology platform, supported by the functional genomics program (FUGE) in the Research Council of Norway. We also thank PA Gagnaire, J Prunier, S Renaut, M Evans, CR Primmer and two anonymous reviewers for their very constructive inputs. The New Brunswick Environmental Trust Fund provided field funding for this project. Research was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) to VB, from the Canadian Research Chair in genomics and conservation of aquatic resources to LB and from funding provided by the Genomics Research and Development Initiative, Department of fisheries and Oceans (DFO), Canada to PTO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Bourret, V., O'Reilly, P., Carr, J. et al. Temporal change in genetic integrity suggests loss of local adaptation in a wild Atlantic salmon (Salmo salar) population following introgression by farmed escapees. Heredity 106, 500–510 (2011). https://doi.org/10.1038/hdy.2010.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.165
Keywords
This article is cited by
-
Microsatellite and mtDNA-based exploration of inter-generic hybridization and patterns of genetic diversity in major carps of Punjab, Pakistan
Aquaculture International (2024)
-
Genetic analyses reveal panmixia in Indian waters and population subdivision across Indian Ocean and Indo-Malay Archipelago for Decapterus russelli
Scientific Reports (2023)
-
Introgressive hybridisation between domestic pigs (Sus scrofa domesticus) and endemic Corsican wild boars (S. s. meridionalis): effects of human-mediated interventions
Heredity (2022)
-
Genetic diversity and population structure analysis of Lateolabrax maculatus from Chinese coastal waters using polymorphic microsatellite markers
Scientific Reports (2021)
-
Variable levels of introgression between the endangered Podarcis carbonelli and highly divergent congeneric species
Heredity (2021)
