
Abstract
CRISPR/Cas9-based therapeutics hold the possibility for permanent treatment of genetic disease. The potency and specificity of this system has been used to target dominantly inherited conditions caused by heterozygous missense mutations through inclusion of the mutated base in the short-guide RNA (sgRNA) sequence. This research evaluates a novel approach for targeting heterozygous single-nucleotide polymorphisms (SNPs) using CRISPR/Cas9. We determined that a mutation within KRT12, which causes Meesmann’s epithelial corneal dystrophy (MECD), leads to the occurrence of a novel protospacer adjacent motif (PAM). We designed an sgRNA complementary to the sequence adjacent to this SNP-derived PAM and evaluated its potency and allele specificity both in vitro and in vivo. This sgRNA was found to be highly effective at reducing the expression of mutant KRT12 mRNA and protein in vitro. To assess its activity in vivo we injected a combined Cas9/sgRNA expression construct into the corneal stroma of a humanized MECD mouse model. Sequence analysis of corneal genomic DNA revealed non-homologous end-joining repair resulting in frame-shifting deletions within the mutant KRT12 allele. This study is the first to demonstrate in vivo gene editing of a heterozygous disease-causing SNP that results in a novel PAM, further highlighting the potential for CRISPR/Cas9-based therapeutics.
Similar content being viewed by others
Introduction
The discovery of a simple endogenous bacterial system for catalytically cleaving double-stranded DNA has revolutionized the field of therapeutic gene editing. The Type II Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/ CRISPR associated protein 9 (Cas9) is a programmable RNA guided endonuclease, which has recently been shown to be effective at gene editing in mammalian cells.1 This highly specific and efficient RNA-guided DNA endonuclease may be of therapeutic importance in a range of genetic diseases. The CRISPR/Cas9 system relies on a single catalytic protein, Cas9 that is guided to a specific DNA sequence by 2 RNA molecules; the tracrRNA and the crRNA.1 Combination of the tracrRNA/crRNA into a single guide RNA molecule (sgRNA)2, 3 has led to the rapid development of gene editing tools potentially specific for any target within the genome. Through the substitution of a 20-nucleotide sequence within the sgRNA, to one complimentary to a chosen target, a highly specific system can be generated in a matter of days. One caveat of this system is that the endonuclease requires a protospacer adjacent motif (PAM), located immediately 3′ of the sgRNA binding site. This PAM sequence is an invariant part of the DNA target but not present in the sgRNA,2, 3 while its absence at the 3′ end of the genomic target sequence results in the inability of the Cas9 to cleave the DNA target.4
The introduction of this technology into a therapeutic setting raised questions over its potential allele-specificity. However, as previously demonstrated by Smith et al. and Yoshimi et al. in animal models, simply altering a single nucleotide within the 20-nucleotide sequence in the sgRNA the CRISPR/Cas9 system renders it unable to bind and cleave the target site.5, 6 This offers the potential of targeting disease causing dominant-negative single-nucleotide polymorphisms (SNPs) to disable the mutant allele, whereas expression of the wild-type allele would remain unaffected.
Using an alternative approach, this study examined the potential of an allele-specific CRISPR/Cas9 system for hereditary corneal dystrophies, specifically focusing on a dominant-negative mutation in KRT12 (encoding keratin 12, K12), Leu132Pro (c. 395 T>C), which results in Meesmann’s epithelial corneal dystrophy (MECD; OMIM:122100).7 Interestingly, this mutation results in the manifestation of a novel Streptococcus pyogenes PAM, not present in the wild-type allele. By incorporating the 20 nucleotides 5′ of this novel PAM into an sgRNA, we sought to establish whether allele-specific cleavage of the mutant allele could be induced. In a heterozygous cell, this double-strand break would either lead to non-homologous end joining (NHEJ), which frequently results in a frameshift and the manifestation of a premature stop codon, or homology-directed repair where recombination with the wild-type allele directs repair of the mutant sequence. In the case of KRT12, both outcomes could be considered a therapeutic success; either expression of the dominant-negative mutant K12 protein is abolished by NHEJ, which is tolerated, as KRT12 has been shown not to demonstrate haploinsufficiency,8 or the mutant allele is repaired by homology-directed repair, resulting in the repair of the K12-Leu132Pro allele.
Through use of techniques adapted from our previous research identifying therapeutic allele-specific short interfering RNAs,7, 9, 10, 11 this Cas9-based gene-editing system was found to be potent and allele specific. This study constitutes the first description of allele-specific CRISPR/Cas9 gene editing at a novel PAM created by a disease-causing heterozygous SNP.
Results
Construction of a KRT12-specific sgRNA
An analysis of the sequence changes that result from MECD-causing KRT12 missense mutations revealed that the L132P mutation7 that causes the severe form of MECD coincidentally results in the generation of a novel PAM site (AAG>AGG). An sgRNA (sgK12LP) complementary to the sequence 20 nucleotides adjacent to the 5′-end of the novel PAM site generated by the KRT12 L132P mutation was designed and assessed for potential off targets using the ‘Optimised CRISPR Design Tool’ provided online by the Zhang lab, MIT 2013, (Figure 1, red). The sgRNA was calculated as having a score of 66% using this system, where a score >50% is deemed to be of high quality with a limited number of predicted possible off targets.

Design of sgRNAs for targeting wild-type and mutant K12 alleles. An sgRNA to use the SNP-derived PAM found on the K12-L132P allele was designed (red). This PAM is absent from the wild-type allele. A second sgRNA to target both wild-type and mutant K12 alleles (green) was also designed and used as a positive control.
Assessment of sgK12LP allele specificity and potency in vitro
The allele-specificity and potency of sgK12LP was assessed in vitro, in HEK AD293 cells, using exogenous expression constructs for wild-type and mutant K12. Allele specificity was first determined using a dual-luciferase reporter assay (Figure 2a). Firefly luciferase activity was found to be significantly decreased in cells expressing either K12WT-Luc or K12LP-Luc and treated with sgK12. A potent and allele-specific reduction of firefly luciferase activity was observed in cells treated with sgK12LP. In cells expressing K12LP-Luc, a reduction of 73.4±2.7% (P<0.001) was observed (Figure 2a). This allele-specific and potent knockdown was also observed by western blotting, in cells expressing either K12WT-HA or K12LP-HA (Figure 2b; image representative of four blots) and quantification by densitometry revealed a significant reduction of 32% in K12LP-HA protein by sgK12LP in comparison with K12WT-HA protein (P<0.05). In cells treated with sgK12, both wild-type and mutant K12 protein was found to have decreased, whereas in those treated with sgK12LP there appeared to be no effect on expression of the wild-type protein but a significant knockdown of the mutant K12 protein (Figure 2b).

Evaluation of allele specificity and potency of sgK12LP using exogenous constructs. Exogenous expression constructs for wild-type and mutant K12 were employed to test the allele-specificity and potency of sgK12LP. (a) A dual luciferase assay demonstrated the allele-specificity of the sgK12LP plasmid, whereas potency was shown to be comparable to that of the sgK12 construct. N=8 (b) Western blotting further demonstrated these attributes with a noticeable reduction in K12-L132P protein in cells treated with sgK12LP in comparison with cells treated but expressing K12 wild-type protein. β-Actin was used as a loading control. (c) Quantitative reverse transcriptase-PCR for total K12 in cells expressing both wild-type and mutant alleles demonstrated a knockdown in mRNA expression. N=4 (d) Allele proportions of this mRNA knockdown were then quantified by pyrosequencing, confirming an allele knockdown of the mutant allele in cells co-expressing both KRT12 alleles and treated with sgK12LP. N=4, *P<0.05, **P<0.01, ***P<0.001.
To support this data, observed at the protein level, quantitative reverse transcriptase-PCR and pyrosequencing were carried out to determine allele specificity and potency at the mRNA level. In cells expressing both wild-type and mutant K12 simultaneously (in a 1:1 expression ratio) and treated with each of the three test Cas9/sgRNA expression constructs (NSC, K12 and K12LP), quantitative reverse transcriptase-PCR was used to determine knockdown of total K12 mRNA (Figure 2c). A potent reduction of 73.1±4.2% (P<0.001) of total K12 mRNA was observed in sgK12-treated cells, with a lesser reduction of 52.6±7.0% (P<0.01) measured in sgK12LP-treated cells (Figure 2c). Pyrosequencing was used to determine the intracellular proportion of the remaining mature mRNA species after treatment with these sgRNAs (Figure 2d). Proportions of mRNA were calculated as ‘percentage of K12-L132P’/percentage of K12-WT’. Cells treated with sgNSC were normalized to 1, assuming a ratio of 1:1 between mutant and wild-type K12 mRNA. In cells tested with sgK12, a K12 mutant mRNA proportion of 0.89±0.03 was observed, but the difference to the NSC control was not significant (P<0.14). In those cells treated with sgK12LP, a K12 mutant mRNA proportion of 0.28±0.02 was detected and was significantly altered in comparison with the sgNSC-treated cells (P<0.001) (Figure 2d).
Determination of the efficacy of sgRNA-K12LP in vivo
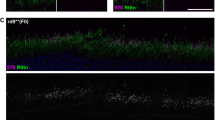
Intrastromal injection of the Cas9-GFP construct resulted in the presence of green fluorescent protein (GFP) protein in the corneal epithelium at 24 h post injection (Figure 3a). Transient expression of GFP was found up to 48 h post injection. Following intrastromal injection of either the sgK12LP or sgNSC expression constructs into K12-L132P humanized heterozygous mice and an incubation period of 48 h, mice were euthanized and genomic DNA (gDNA) prepared from the corneas. gDNA from the corneas of four sgK12LP- or sgNSC-treated animals was pooled and PCR amplification of exon 1 of the humanized K12-L132P gene, cloning and sequencing was performed. Of 10 clones established from gDNA of eyes treated with sgNSC, the K12-L132P sequence remained intact in all 10. Thirteen individual clones from sgK12LP-treated eyes were sequenced; eight were found to contain an unaltered KRT12 L132P human sequence, whereas five clones demonstrated NHEJ around the predicted cleavage site of the Cas9/sgK12LP complex (Figure 3b). In one clone (1), an insertion of 1 nucleotide was found, with a deletion of 32 nucleotides. Large deletions of up to 53 nucleotides were observed in vivo (clone 5). Of these 5 clones, 4 contained deletions (clones 1 and 3–5) that are predicted to result in a frameshift that would lead to the occurrence of an early stop codon. The top 2 predicted exonic off-target sites of sgK12LP in mouse were also assessed using this method. Ten clones were sequenced for each target and none were found to have undergone nonspecific cleavage.

Demonstration of sgK12LP-induced NHEJ in vivo. GFP expression was observed in the corneal epithelium of mice at 24 h post intrastromal injection, demonstrating the efficacy of an intrastromal plasmid injection for transfecting the corneal epithelium (a; N=2). No GFP expression was observed 48 h post injection. Sequencing of the gDNA from human K12-L132P heterozygous mice injected with the sgK12LP construct demonstrated large deletions and the induction of NHEJ due to cleavage of the KRT12-L132P allele. Of 13 clones sequenced, 5 were found to have undergone NHEJ (b).
Discussion
Since the discovery of the bacterial CRISPR/Cas9 system and its proven efficacy in mammalian cells, a number of studies have demonstrated the potential therapeutic value of this system for a wide range of genetic diseases.12, 13, 14 This technology has been further used in an allele-specific manner, demonstrating the potential of the CRISPR/Cas9-based system for treating dominant hereditary conditions.5, 6 Within this study, we aimed to develop an allele-specific genome-editing system employing S. pyogenes Cas9 for the KRT12-L132P mutation that leads to the onset of MECD. However, this research took a novel approach in targeting a heterozygous mutation that results in the occurrence of a novel PAM site in the mutant allele, caused by the single base change at position 395 (T>C). The aim of this study was to determine whether targeting the sequence adjacent to this PAM site could result in potent and allele-specific cleavage of the KRT12-L132P allele, establishing the potential for therapeutic utilization of these SNP-derived PAMs for dominantly inherited diseases.
An sgRNA adjacent to this SNP-derived PAM was assessed by co-transfection of Cas9/sgRNA expression constructs, with exogenous reporter constructs in mammalian cells. In the event of sgRNA recognition of its target site and Cas9-mediated cleavage of the exogenous luciferase reporter construct, luciferase mRNA transcription is disrupted and the plasmid becomes functionally silent. Neither NHEJ nor homology-directed repair need occur to observe this effect. Through utilization of dual-luciferase reporter assays, western blotting, quantitative reverse transcriptase-PCR and pyrosequencing the expression levels of wild-type and mutant K12 were quantified at both the mRNA and protein level. The Cas9/sgRNA complex using the SNP-derived PAM was found to be highly potent, similar to that of a positive control KRT12 sgRNA common to both wild-type and mutant alleles, and allele-specific, as demonstrated by no observable knockdown of wild-type K12 expression.
To evaluate the targeting ability of the Cas9/sgRNA complex in vivo and to identify functional knockout of the mutant allele by NHEJ, a mouse model heterozygous for the human KRT12-L132P allele was injected intrastromally with the allele-specific Cas9/sgRNA expression construct. Corneal gDNA was extracted and the human KRT12-L132P allele was PCR amplified, cloned and sequenced. Of the clones sequenced, 38.5% were found to have undergone NHEJ in vivo. Deletions ranging from 18 to 53 nucleotides were observed. As previous research into K12 has demonstrated, a 50% reduction in the expression of the mutant allele would suffice in alleviating the phenotype,15 whereas ablation of one KRT12 allele does not negatively affect corneal integrity.8 We will endeavor to increase the targeting efficiency of this system through methods including in-vivo electroporation.16 These studies will also investigate the corneal integrity of heterozygous mice, having undergone Cas9-mediated NHEJ, over a prolonged period of time.
This research demonstrates the potential for exploitation of these SNP-derived PAMs in the development of personalized therapeutics for dominantly inherited conditions. A review of dystrophic mutations in KRT3, KRT12, TGFBI and COL8A2 showed that of the 76 known missense mutations, 27 (36%) resulted in the manifestation of a novel PAM (Supplementary Table S1), again supporting the potential to use these SNP-derived PAMs in the advancement of novel gene therapies. Following genetic diagnosis, a personalized sgRNA expression vector can be designed and synthesized in relation to a patient’s particular disease-causing mutation within a matter of weeks. Within this study we have focused on corneal dystrophies but the potential exists for developing therapies for a wide range of heterozygous disease-causing SNPs, whether to knockout the mutant allele, as demonstrated here using NHEJ, or to repair the mutant allele, using similar methods already published using homology-directed repair.17
Materials and methods
Constructs
Three plasmids expressing Cas9 and an sgRNA were used throughout all experiments. The non-targeting plasmid used was pSpCas9(BB)-2A-Puro (PX459), a gift from Professor Feng Zhang (Broad Institute, MIT; Addgene plasmid 48139).17 Following a published protocol,17 the plasmid containing the sgRNA specific to the K12-L132P allele was designed by annealing and cloning the following 2 primers (Life Technologies, Paisley, UK): 5′-CACCGTAGGAAGCTAATCTATCATT-3′ and 5′-AAACAATGATAGATTAGCTTCCTAC-3′ into pSpCas9(BB)-2A-Puro. This sgRNA corresponds to the 20 nucleotides directly 3′ of the allele-specific PAM found on the K12-L132P allele (Figure 1, red), hereafter named sgK12LP. A Cas9/sgRNA plasmid to target both wild-type and mutant K12 sequences was constructed (Sigma, Gillingham, UK) and used as a positive control (Figure 1, green).
Additional K12 expression constructs previously described7 were used to assess allele specificity and potency. Firefly luciferase plasmids with the full mRNA sequence for either K12-WT or K12-L132P inserted 3′ of the stop codon, hereafter named as K12WT-Luc and K12LP-Luc, respectively,7 and expression plasmids for mature haemagglutinin (HA)-tagged K12-WT and K12-L132P protein,10 with plasmids hereafter known as K12WT-HA and K12LP-HA, respectively, were used. An expression construct for Renilla luciferase (pRL-CMV, Promega, Southampton, UK) was used for the dual-luciferase assay to normalize transfection efficiency.
Dual-luciferase assay
A dual-luciferase assay was used to quantify potency and allele-specificity of the three test sgRNAs in exogenous constructs, using methods adapted as previously described.9, 11, 18 In short, HEK AD293 cells (Life Technologies) were transfected using Lipofectamine 2000 (Life Technologies) with both K12WT-Luc or K12LP-Luc expression constructs and sgNSC, sgK12 or sgK12LP constructs at a ratio of 1:4. Cells were incubated for 72 h before being lysed and the activities of both Firefly and Renilla luciferase quantified. In all, eight replicates were carried out for each transfection condition.
Western blotting
HA-tagged wild-type (K12WT-HA) and mutant (K12LP-HA) expression constructs7 were transiently co-transfected with each of the sgRNAs at a ratio of 1:4 into HEK AD293 cells in duplicate using Lipofectamine 2000 (Invitrogen), using similar methods as previously described.9 Transfected cells were incubated for 72 h. Expression of HA-tagged K12 and β-actin was analysed using a rabbit polyclonal antibody to HA (Abcam, Cambridge, UK; ab9110, 1:2000) and a mouse monoclonal antibody to human β-actin (Sigma, 1:15 000) using standard methods.9 Membranes were incubated with a secondary horseradish peroxide-conjugated polyclonal swine anti-rabbit antibody (DakoCytomation, Ely, UK) or a horseradish peroxide-conjugated goat anti-mouse antibody (DakoCytomation), respectively. Protein binding was detected by standard chemiluminescence (Life Technologies). Densitometry was performed using ImageJ,19 to quantify the band intensity of the HA-tagged K12 (n=4). This was normalized to the band intensity of β-actin.
Quantitative real-time PCR
Transfections were carried out in the same manner as described for western blotting; however, both K12WT-HA and K12LP-HA were simultaneously transfected into cells. All transfections were carried out in triplicate. Following transfection, cells were incubated for 48 h and RNA extracted using the RNAeasy Plus kit (Qiagen, Venlo, The Netherlands). Following cDNA conversion of 500 ng of RNA (Life Technologies) quantitative real-time PCR was performed to quantify levels of KRT12 mRNA. A KRT12 assay was used (assay Id 140679; Roche, West Sussex, UK) alongside an HPRT assay (assay ID 102079; Roche) and a GAPDH assay (assay ID 141139; Roche). Each sample was run in triplicate for each assay and relative gene expression was calculated using the ΔΔCT method.20 KRT12 expression levels were normalized against HPRT and GAPDH, where expression of both reference genes was deemed to be ‘stable’ across treatment groups, using the BestKeeper software tool.21
Pyrosequencing
Using the same cDNA samples assessed by quantitative reverse transcriptase-PCR, pyrosequencing was carried out to determine the ratio of remaining K12-L132P mRNA to K12-WT mRNA, exactly as described previously.10
KRT12 transgenic mouse
A C57 mouse model was obtained, with a human K12-L132P allele knocked in to replace the endogenous mouse Krt12 coding sequence (details on request). This allowed for the in-vivo targeting of KRT12-L132P by the allele-specific sgRNA and Cas9. Female heterozygous mice at 24 weeks old were used, where one copy of the human K12-L132P allele and one copy of murine Krt12 were present. Standard PCR and Sanger dideoxynucleotide sequencing was used to genotype the mice and confirm heterozygosity of the K12-L132P allele. Randomization of animals was not required, as this study investigated the effect of treatment on one cornea, whereas the other cornea of the same animal was used as the negative control. Investigators were not blinded in this study. All experimentation complied with ethical regulations and was approved by the local ethics committee.
In-vivo intrastromal ocular injection
To achieve transient expression of the allele-specific sgRNA and the Cas9, the sgK12LP plasmid was introduced into the corneal stroma of the heterozygous knock-in mice by intrastromal ocular injection, following previously described protocols.22 To assess this delivery method, wild-type mice were first injected with 4 μg of a Cas9-GFP plasmid (pCas9D10A_GFP), a gift from Kiran Musunuru (Addgene plasmid 44720). Mice were culled at 24, 48 and 72 h, and corneas fixed in 4% paraformaldehyde and processed using standard histological procedures. Five-micrometre-thick sections were cut, rehydrated and imaged by fluorescent microscopy. Mice were administered with general anaesthetic and a local anaesthetic to the cornea. A qualified ophthalmologist injected 4 μg of sgK12LP or sgNSC plasmid diluted in a total of 3 μl phosphate-buffered saline into the cornea of the right eye and the left eye, respectively, of four mice. Mice were culled 48 h post treatment.
Sequencing and determination of NHEJ
Once the mice were culled, the eyes were enucleated and the corneas were dissected. gDNA was extracted using a DNA extraction kit (Qiagen) and samples were pooled into two treatment groups: sgK12LP and sgNSC. Samples underwent PCR amplification using the following two primers to amplify the region around the K12-L132P mutation: 5′-ACACCCATCTTGCAGCCTAT-3′ and 5′-AAAATTCCCAAAGCGCCTC-3′. PCR products were gel purified and ligated into the CloneJet cloning vector (Life Technologies) and were used to transform DH5α competent cells (Life Technologies). A total of 13 clones were selected and plasmid DNA prepared using a miniprep kit (Qiagen) following manufacturer’s procedures. DNA from the 13 clones was then sequenced (Department of Zoology, University of Oxford) using the sequencing primers provided with the CloneJet vector. The two most likely exonic off-target sites for sgK12LP in the mouse genome, as predicted by the Zhang Lab online tool (http://crispr.mit.edu) were assessed in the same way, where 10 colonies were selected for analysis for each predicted off target. The predicted off-target sites were 5′-TAAGTAGCTGATCTATCAGTGGG-3′ (Gon4l) and 5′-TGGGAAGCATATCTGTCATTTGG-3′ (Asphd1). Only these two sites were selected, as they were the only two to have a calculated off-target score >0.1.
Statistics
All error bars represent the s.e.m. unless stated otherwise. Significance was calculated using an unpaired t-test, as all samples demonstrated the same distribution. Statistical significance was set at 0.05%. Variance was calculated among groups and deemed to be similar.
References
Hsu PD, Lander ES, Zhang F . Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014; 157: 1262–1278.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014; 343: 84–87.
Wang T, Wei JJ, Sabatini DM, Lander ES . Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014; 343: 80–84.
Westra ER, Semenova E, Datsenko KA, Jackson RN, Wiedenheft B, Severinov K et al. Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet 2013; 9: e1003742.
Smith C, Abalde-Atristain L, He C, Brodsky BR, Braunstein EM, Chaudhari P et al. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol Ther 2014; 23: 570–577.
Yoshimi K, Kaneko T, Voigt B, Mashimo T . Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR-Cas platform. Nat Commun 2014; 5: 4240.
Liao H, Irvine AD, Macewen CJ, Weed KH, Porter L, Corden LD et al. Development of allele-specific therapeutic siRNA in Meesmann epithelial corneal dystrophy. PLoS One 2011; 6: e28582.
Kao WW, Liu CY, Converse RL, Shiraishi A, Kao CW, Ishizaki M et al. Keratin 12-deficient mice have fragile corneal epithelia. Invest Ophthalmol Vis Sci 1996; 37: 2572–2584.
Courtney DG, Atkinson SD, Moore JE, Maurizi E, Serafini C, Pellegrini G et al. Development of allele-specific gene-silencing siRNAs for TGFBI Arg124Cys in lattice corneal dystrophy type I. Invest Ophthalmol Vis Sci 2014; 55: 977–985.
Courtney DG, Atkinson SD, Allen EHA, Moore JE, Walsh CP, Pedrioli DML et al. siRNA silencing of the mutant keratin 12 allele in corneal limbal epithelial cells grown from patients with Meesmann’s epithelial corneal dystrophy. Invest Ophthalmol Vis Sci 2014; 55: 3352–3360.
Allen EHA, Atkinson SD, Liao H, Moore JE, Leslie Pedrioli DM, Smith FJD et al. Allele-specific siRNA silencing for the common keratin 12 founder mutation in Meesmann epithelial corneal dystrophy. Invest Ophthalmol Vis Sci 2013; 54: 494–502.
Schwank G, Koo B-K, Sasselli V, Dekkers JF, Heo I, Demircan T et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013; 13: 653–658.
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 2014; 32: 551–553.
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S et al. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013; 13: 659–662.
Cao T, Longley MA, Wang X, Roop DR . An inducible mouse model for epidermolysis bullosa simplex: implications for gene therapy. J Cell Biol 2001; 152: 651–656.
Blair-Parks K, Weston BC, Dean DA . High-level gene transfer to the cornea using electroporation. J Gene Med 4: 92–100.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F . Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8: 2281–2308.
Atkinson SD, McGilligan VE, Liao H, Szeverenyi I, Smith FJD, Moore CBT et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Invest Dermatol 2011; 131: 2079–2086.
Schneider CA, Rasband WS, Eliceiri KW . NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9: 671–675.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP . Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett 2004; 26: 509–515.
Moore JE, McMullen CBT, Mahon G, Adamis AP . The corneal epithelial stem cell. DNA Cell Biol 21: 443–451.
Acknowledgements
We acknowledge Professor Irwin McLean for providing the MECD mouse model used within and Professor Feng Zhang for providing the Cas9/sgRNA expression plasmid. This work was supported by Fight for Sight UK (1880 and 1450/1451), the Medical Research Council (G0801742 and G0802780) and The Belfast Association for the Blind.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Courtney, D., Moore, J., Atkinson, S. et al. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther 23, 108–112 (2016). https://doi.org/10.1038/gt.2015.82
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2015.82
This article is cited by
-
The application and progression of CRISPR/Cas9 technology in ophthalmological diseases
Eye (2023)
-
Detailed corneal and genetic characteristics of a pediatric patient with macular corneal dystrophy - case report
BMC Ophthalmology (2021)
-
Engineered dual selection for directed evolution of SpCas9 PAM specificity
Nature Communications (2021)
-
Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects
Signal Transduction and Targeted Therapy (2020)
-
Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities
Nature Communications (2020)
