
Abstract
Influenza virus (IV) infection is a major public health problem, causing millions of cases of severe illness and as many as 500 000 deaths each year worldwide. Given the limitations of current prevention or treatment of acute influenza, novel therapies are needed. RNA interference (RNAi) through microRNAs (miRNA) is an emerging technology that can suppress virus replication in vitro and in vivo. Here, we describe a novel strategy for the treatment of infuenza based on RNAi delivered by a replication-defective adenovirus (Ad) vector, derived from chimpanzee serotype 68 (AdC68). Our results showed that artificial miRNAs (amiRNAs) specifically targeting conserved regions of the IV genome could effectively inhibit virus replication in human embryonic kidney 293 cells. Moreover, our results demonstrated that prophylactic treatment with AdC68 expressing amiRNAs directed against M1, M2 or nucleoprotein genes of IV completely protected mice from homologous A/PR8 virus challenge and partially protected the mice from heterologous influenza A virus strains such as H9N2 and H5N1. Collectively, our data demonstrate that amiRNAs targeting the conserved regions of influenza A virus delivered by Ad vectors should be pursued as a novel strategy for prophylaxis of IV infection in humans and animals.
Similar content being viewed by others


Introduction
Influenza A virus is a highly contagious pathogen that can infect a variety of hosts, including poultry and human. On average, seasonal influenza virus (IV) causes 3–5 million human illness and 250 000–500 000 human deaths annually.1 The outbreak of high pathogenic avian influenza H5N1 in Hongkong, which was first observed in 1997, raised public attention to human infection with avian IVs.2 In March 2013, H7N9 avian IV was first isolated in patients presenting with rapidly progressing lower respiratory tract infections in China.3 Up to April 2014, a total of 402 H7N9 cases have been reported, of which 146 died.4 A third avian virus, H10N8, termed A/Jiangxi-Donghu/346/2013, was reported in December of 2013. The virus infected a 73-year-old woman in the Jiangxi province of China, who subsequently died.5 A second human case was recorded in January of 2014 from Nanchang.6 The infections with novel strains to which vaccines are not available require more effective ways to prevent influenza.
Individuals can be protected from IV infection by vaccination.7 One commonly used influenza vaccine on the market is an annually reformulated trivalent split vaccine. It is composed of two strains of influenza A virus and one or two influenza B virus strains, selected based on the strains that are predicted to be prevalent in the upcoming influenza season.7 This vaccine confers up to 80% effective protection to healthy adults but performs poorly in the elderly. As protection is linked to induction of strain-specific neutralizing antibodies, the efficacy of trivalent split vaccine is markedly reduced in years when the vaccine strains mismatch those of the predominant strains.8 Thus, reagents, which provide cross immunity against a broad spectrum of viruses, are in critical demand.
RNA interference (RNAi) is a widely applied strategy in inhibiting sequence-specific gene expression by messenger RNA degradation or translation inhibition. It can be initiated by double-strand RNA (dsRNA) and regulated by small RNA, including small interfering RNAs and microRNAs (miRNA).9 This technology has been extended to prevention of viral infections, including those with HIV,10 hepatitis C virus11 and IV.12 Ge et al.13, 14 and Tompkins et al.8 have reported small interfering RNAs targeting conserved regions of the IV genome could potently inhibit replication of different strains both in vitro and in vivo. However, small interfering RNAs can compete with endogenous miRNA for gene-silencing components, which limits its clinic application.15 Artificial miRNA (amiRNA), on the other hand, is less toxic with comparative silencing efficiency and therefore provides an improved antiviral tool. Furthermore, in case of an unexpected outbreak, traditional vaccines require several days to achieve protective titers of antibodies. Their efficacy in preventing disease in already exposed individuals is thus expected to be limited; RNAi functions shortly after being introduced into cells and could thereby provide benefits to already infected individuals as remains to be explored.
Here, we developed a platform by using a recombinant adenovirus (Ad) vector to deliver amiRNA prior to infection of mice with lethal doses of influenza A viruses. The Ad vector expressed amiRNA targeting matrix (M) or nucleoprotein (NP) genes of H1N1. These recombinant Ads could potently suppress influenza replication both in vitro and in vivo. More importantly, one single dose of the Ad vector given intranasally completely protected ICR mice from homotypic H1N1 challenge and provided partial protection against heterosubtypic H9N2 or H5N1 challenge.
Results
Design and screen for amiRNAs targeting influenza A virus
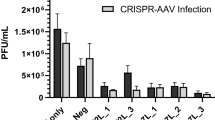
It has been widely reported that amiRNA can potently suppress viral replication with minimal toxicity.16, 17, 18 Here we choose the highly conserved NP, M1 and M2 genes of A/PR8 virus as amiRNA targets for antiviral treatment by silencing genome expression. A total of 11 amiRNAs including M1-89, M1-420, M1-510, M1-627, M2-83, M2-117, M2-118, M2-235, NP-490, NP-856 and NP-969 were designed. The sequences of the pre-artificial miRNA are shown in Supplementary Table S1. To assess the ability of the amiRNAs to inhibit virus replication, we established a screen system in human embryonic kidney 293 T (HEK 293 T) cells. As shown in Figure 1a, pretreatment of HEK 293 T cells with influenza-specific amiRNAs reduced viral copies markedly compared with those in mock-treated cells (P<0.0001). Especially amiRNAs M1-89, M2-117, NP-490, NP-858 and NP-969 inhibited viral replication by >80%.

Screen for functional influenza-specific artificial microRNA. Both single (a) and chained (b) amiRNA candidates (1 μg) were transiently transfected into 293 T cells (plated at 1 × 105 per well in 24-well plate on the previous day), which were challenged 24 h later with 0.0005 MOI A/PR8 for screening by real-time PCR. pcDNA 6.2-GW/miR-neg transfectant and virus only served as controls, respectively. P<0.0001 for all amiRNAs compared with either controls. To assess the inhibitory effect on polymerase activity, indicated dose of NP-specific amiRNA plasmids along with mini-genome reporter assay-related plasmids were transfected into 293 T cells (c). Firefly luciferase activity were assayed in cell lysates and compared with Renilla activity 24 h later. Control group was normalized to 100%. NP-856, NP-969, M1-89+NP-856 and M1-89+NP-969 were in comparison with the control (P<0.05). All data were repeated in triplicate. One-way ANOVA was applied to compare with the control group.
To assess whether we could enhance inhibition of viral replication, we co-expressed the most potent amiRNAs M1-89, M2-117, NP-856 and NP-969 and tested them for inhibition of A/PR8 replication. Virus inhibition could not be further enhanced in HEK 293 T cells treated with the chained amiRNAs (Figure 1b) (P<0.0001). Overall, these results indicated that IV replication could be significantly repressed by specific amiRNAs treatment in vitro, and chaining different amiRNAs failed to increase the inhibitory effects. One drawback of RNAi-mediated antiviral strategies is loss of targets in viral mutants,19 amiRNAs targeting several genes of IV may provide better viral suppression against such mutants.
NP-specific amiRNAs inhibit the RNP activity
During the IV life cycle, NP associates with PB2, PB1 and PA and forms the ribonucleoprotein (RNP) complex, which is indispensable for virus replication and transcription. Therefore, we hypothesized that NP-specific RNAi could not only contribute to target protein degradation, but also decrease viral RNA accumulation by inhibiting RNP activity. To assess this hypothesis, we constructed a luciferase-based mini-genome reporter assay system as previously described.20 Luciferase activity was evaluated 24 h post transfecion. Mock control was normalized to 100%. As shown in Figure 1c, the relative luciferase activity was significantly suppressed in a dose-dependent manner in the amiRNA-expressing cells (P<0.05). Notably, cultures treated with high doses of the amiRNAs showed 70–80% inhibition of RNP activity.
Ad vector-delivered amiRNAs inhibit IV replication in A549 cells
For in vivo prevention of influenza infection, it is essential to effectively deliver amiRNAs into the site of virus infection, that is, the respiratory tract.21 We choose Ad vectors as delivery system considering its broad tissue tropism and effective expression of foreign genes within cells of the lower respiratory tract.22 To test the antiviral activity of these Ad vector-encoded amiRNAs, A549 cells were transduced with Ad-amiRNA at different doses and then infected with 0.001 MOI of A/PR8 virus 24 h later. Cells treated with AdC68-miR-empty vector or non-transduced cells served as controls. In Ad-amiRNAs-treated cells, NP expressions were significantly suppressed. Level of suppression was linked to the Ad vector dose (Figure 2a). Remarkably, rAd(NP-969) and rAd(M1-89+NP-969) nearly abolished NP expression at the highest 1010 virus particle (vp) dose. The inhibition difference was mainly due to different amiRNAs rather than different expression level as assessed by small RNA northern blot (data not shown). To further analyze vector-derived amiRNA-mediated attenuation, we determined the M2 protein level at 12 h, 24 h and 36 h post influenza challenge. As shown in Figure 2b, Ad-amiRNA treatment significantly reduced viral protein over time.

Influenza-specific Ad-amiRNAs downregulated viral protein in a dose-dependent manner. Expression of NP in different Ad-amiRNAs treated A549 cells was determined 24 h post influenza challenge in comparison with beta-actin (a). To analyze amiRNA-mediated attenuation, the M2 protein level was assessed in A549 cells (treated with 1010 vp of indicated Ad-amiRNAs on the previous day) at 12 h, 24 h and 36 h post influenza challenge (b). NC, negative control; VC, irrelative virus control.
Ad-amiRNA vectors reduce IV replication and alleviate associated disease in mice
ICR mice were inoculated intranasally with 1011 vp of Ad-amiRNA vectors and challenged 24 h later with five lethal dose (LD)50 of A/PR8 virus. AdC68-miR-empty served as a control. Histological changes were assessed on sections of the left upper lung lobes of each mouse 5 days post challenge by hematoxylin and eosin staining. Control mice developed perivascular and interstitial infiltrates with an average pathogenic score of 4.7. Vaccinated mice presented with less pronounced inflammatory responses (Figures 3a and b). Consistent with histological results, viral RNA copies determined from the right inferior lung lobes of individual mice were significantly lower in Ad-amiRNA vectors treated mice compared with control mice (Figure 3c) (rAd(NP-856), P=0.017, rAd(M1-89+NP-856), P=0.005, others, P=0.002).

Influenza targeting Ads protected ICR mice completely from lethal challenge of A/PR8. ICR mice (n=15) vaccinated with indicated Ads were challenged 24 h later with 5LD50 of A/PR8. Five mice from each group were euthanized and dissected 5 d.p.i. Left upper lung lobes were stained with hematoxylin and eosin (a) and scored for histological changes (b) (rAd(NP-856), P=0.011, rAd(M1-89+NP-856), P=0.001, others, P<0.0001). Sections were representative of each group. Lung viral copy numbers were determined by absolute quantification RT-PCR (c) (rAd(NP-856), P=0.017, rAd(M1-89+NP-856), P=0.005, others P=0.002). All data were repeated in triplicate. Remaining 10 mice of each group were monitored 21 days for weight loss (d) and survival rates (e). P-value was 0.006 for rAd(NP-856) and <0.0001 for other Ads in survival experiment (e). Mean weight+s.d. was determined at each time point. One mouse survived in control group whose weight loss was not included. One-way ANOVA was applied to compare virus titers and pathology grade. χ2-test was performed to compare the survival rates.
To ensure that inhibition of virus replication had clinic benefits, we monitored weight loss of additional mice for 21 days. As depicted in Figures 3d and e, all of the control mice lost 20–30% of their original weight after challenge. Most of the mice treated with IV-specific amiRNAs initially gained weight, which they then by day 5–7 started to lose. Weight lost by treated mice varied depending on the Ad vector treatment. Within the vaccine groups, mice treated with rAd(M1-87+NP-969) did not lose any weight, whereas mice treated with rAd(NP-856) lost the most weight (>15%). Weight loss of IV-specific amiRNA mice peaked by 7–9 days after challenge and then mice regained weight gradually. Mice from control group continued to lose weight till they died or required euthanasia. Overall mice treated with most of the Ad vectors expressing IV-specific amiRNAs were completely protected from death (P<0.0001), whereas mice treated with rAd(M2-117) (P<0.0001) or rAd(NP-856) (P=0.006) showed significantly enhanced survival compared with controls.
Influenza NP induces potent CD8+ T-cell responses in mice.23, 24 We expected that knockdown of NP would reduce this response by lowering antigenic loads. Thus, we performed NP-specific tetramer staining and intracellular cytokine staining on blood-derived lymphocytes from rAd(NP-969)-treated mice using tetramer stains and intracellular cytokine staining for IFN-γ (Figure 4). Here, we challenged mice with 1LD50 of A/PR8, so that all mice could survive until 4 weeks. Both responses were significantly reduced when tested 2 and 4 weeks after influenza A virus challenge. These results again supported the notion that Ad vector-delivered amiRNAs significantly reduced IV replication.

NP-specific Ad-amiRNAs downregulated CD8+ T-cell response via NP degradation. C57Bl/6 mice (n=5) were immunized with 1 × 1011 vp of Ads-amiRNAs and challenged with 1LD50 of A/PR8 24 h later. Frequency of NP-specific CD8+ T cells (a) or IFN-γ+CD8+ T cells (b) in peripheral blood of C57Bl/6 mice collected 2 or 4 weeks after infection were determined by tetramer staining or intercellular cytokine staining, respectively (one-way ANOVA).
Cross-protection against heterotypic strains of influenza A viruses
We next assessed whether Ads-amiRNAs afforded cross-protection against heterotypic strains of influenza A virus. Avian IVs have become of particular interest in the past decades because of their potential to infect humans, gain human-to-human transmissibility and then unleash a pandemic.3 Therefore, the above-described mouse experiments were repeated using a low pathogenic avian influenza H9N2 strain for challenge. As shown in Figures 5a and b, inflammation within lungs of Ad-amiRNA-treated mice were markedly reduced with mean scores ranging from 3.2 to 3.8 compared with controls (mean pathology score of control group was 4.9) (P<0.05). Mean viral RNA copy numbers of treated mice were decreased by 102 to 103 fold compared with controls (P<0.0001) (Figure 5c). All groups exhibited significant weight loss after H9N2 challenge (Figure 5d), among which, rAd(NP-969) treated mice showed the least weight loss (around 10%). Furthermore, all mice in rAd(NP-969) group survived after the H9N2 lethal challenge (P<0.0001), whereas rAd(M1-89), rAd(M1-89+NP-969) and rAd(M1-89+M2-117) provided 40–70% protection. All of the mice treated with other Ad vectors including the control vector succumbed (Figure 5e).

Influenza targeting Ads protected ICR mice partially from lethal challenge of H9N2. ICR mice (n=15) vaccinated with indicated Ads were challenged 24 h later with 5LD50 of H9N2. Five mice from each group were euthanized and dissected 5 d.p.i. Left upper lung lobes were stained with hematoxylin and eosin (a) and scored for histological changes (b). Lung viral copies were determined by absolute quantification RT-PCR (c) (P<0.0001). All data were repeated in triplicate. Remaining 10 mice of each group were monitored 21 days for weight loss (d) and survival rates (e) (survival experiment, rAd(NP-969), P<0.0001, rAd(M1-89), P=0.043, rAd(M1-89+M2-117), P=0.002, others, P=0.005). One-way ANOVA was applied to compare viral copies and pathology grade. χ2-test was performed to compare the survival rates.
Two Ad-amiRNAs were further tested their efficacy against high pathogenic avian influenza H5N1 virus challenge. Reduction in inflammation and viral genome copies were evident in rAd(M1-89+M2-117) and rAd(NP-969) treated mice (Figures 6a and c) with survival rates of 70% (P=0.085) and 60% (P=0.035), respectively as compared with 20% survival of controls (Figure 6e). Weight loss was not evident in both vaccinated groups (Figure 6d). Thus, one single dose of specific amiRNA treatment partially protected against heterotypic challenges.

Influenza targeting Ads protected ICR mice partially from lethal challenge of H5N1. ICR mice (n=15) vaccinated with indicated Ads were challenged 24 h later with 5LD50 of H5N1. Five mice from each group were euthanized and dissected 5 d.p.i. Left upper lung lobes were stained with hematoxylin and eosin (a) and scored for histological changes (b) (P<0.0001). Lung viral RNA copies were determined by absolute quantification RT-PCR (c). P-value was 0.027 for both Ads. All data were repeated in triplicate. Remaining 10 mice of each group were monitored 21 days for weight loss (d) and survival rates (e) (survival experiment, rAd(M1-89+M2-117), P=0.085, rAd(NP-969), P=0.035). One-way ANOVA was applied to compare viral copies and pathology grade. χ2-square test was performed to compare the survival rates.
Duration of protection
To determine the duration of protection, we immunized groups of mice (n=10) with 1011 vp of rAd(NP-969) and challenged them with 5LD50 of A/PR8 virus 1, 5, 7, 10, 15, 30 days later. We observed that the IV-specific Ad vectors could provide complete protection for 5 days (P<0.0001) and partial protection (60–80%) for 15 days (7d post treatment P=0.003, 10 d post treatment P=0.001, 15 d post treatment P=0.029). However, survival rate dropped to 30% when mice were challenged 30 days later, this level of survival was not significantly different from that of control mice (P=0.291) (Figures 7a and b).

Protective duration induced by rAd(NP-969). rAd(NP-969) inoculated ICR mice (n=10) were infected i.n. with 5LD50 A/PR8 at different time points. Weight loss (a) and survival rate (b) were monitored for 21 days post challenge. In survival experiment, P-value was 0.003 for 7d p.t, 0.001 for 10 d p.t, 0.029 for 15 d p.t, 0.291 for 30d p.t and <0.0001 for the rest (χ2-test) (p.t, post treatment).
Discussion
Prediction-based traditional influenza vaccines have several drawbacks. Most notably is lack of cross-protection against antigenic variant strains. Production of some strains in chicken eggs or cell culture is hampered by poor yields. Furthermore, especially vulnerable individuals such as the elderly or humans with immune deficiency fail to respond or only respond poorly to vaccination. Various strategies have been adopted to develop a universal influenza vaccine to achieve better protection against newly evolving strains or viral mutants. For example, M2e conjugate vaccines have been reported to restrict H1N1, H3N1 viral shedding in mice, ferrets and rhesus monkeys.24 A modified vaccinia virus Ankara vector encoding NP and M1 could boost cross-reactive T-cell responses against different subtypes in phase I clinical trial.25 Vaccine including those that achieve cross-protection against most IV strains require several days till protective titers of antibodies are produced. Thus, during an unexpected outbreak, novel treatment options, which very rapidly provide protection are urgently needed. Such treatments can function to complement traditional vaccines and are particularly valuable for individuals known to respond poorly to vaccination. To this end, we decided to directly target influenza viral genome by RNAi.
RNAi might complement and improve traditional tools to control the spread of IVs because of its high specificity, rapid onset of function and low toxicity. Many studies have shown that RNAi can significantly suppress gene expression when delivered into mammalian cells, which raised the possibility that miRNA could inhibit viral gene expression and protect cells from viral replication.10, 11, 12 Application of amiRNA as in vivo prophylaxis vaccines requires safe and effective delivery systems. Previous mouse studies used hydrodynamic in vein injection26 or polyethylenimine vehicles25 to deliver amiRNAs. In this study, we used replication-defective Ad vectors for delivery. Ad vectors are commonly used for gene transfer because of their broad tropism, high transduction efficiency and relatively low reactogenicity. We used a chimpanzee-derived Ad vectors to which most humans fail to have neutralizing antibodies, which could dampen transduction efficacy in humans or eventual target species.22
Our study describes, for the first time, that Ad vector-delivered amiRNAs inhibit IV replication in vitro and in vivo and thereby protecting mice against an otherwise fatal influenza A virus infection. Mice were not only protected against disease following infection with the homologous virus but also against two heterotypic viruses that currently circulate in poultry. Protection, which could be achieved with amiRNAs targeting NP or M genes, was sustained for ~2 weeks; we assume that by then the adaptive immune system of the treated mice had largely eliminated Ad vector antigen-expressing cells. Expressing several amiRNAs by one vector in form of so-called chained amiRNAs failed to show enhanced inhibition of viral replication in vitro or to increase the breadth of protection against heterologous IV challenge in vivo.
Our results pave the way toward exploration of amiRNAs as a possible prophylaxis treatment to curtail IV infection. Additional studies are needed to assess if this strategy can also lessen the impact of already ongoing infections.
Materials and methods
Artificial miRNA design and generation of plasmids miRNA
The artificial miRNAs targeting NP, M1 and M2 gene of the A/Puerto Rico/8/1934 (H1N1) (A/PR8) virus were designed by BLOCK-iT Pol II miR RNAi Designer (http://rnaidesigner.invitrogen.com/rnaiexpress/). Eleven of the most suited sequences were generated and synthesized by Invitrogen (Shanghai, China). Single-stranded oligonucleotides were annealed and further cloned into miRNA expression vector pcDNATM6.2-GW/miR (Invitrogen, Carlsbad, CA). To express multiple amiRNAs co-cistronically, single amiRNA was chained together via enzyme digestion and ligation according to the manufacturer’s instruction. In brief, pre-miRNA inserts were excised by a combination of BamHI and XhoI and cloned into BglII and XhoI predigested backbone vectors.
Cell culture
Human embryonic kidney 293 (HEK 293), HEK 293 T and human lung adenocarcinoma epithelial cell line (A549) were maintained in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum with penicillin (100 U ml−1) and streptomycin (100 μg ml−1).
Cell transfection
All plasmid DNA transfections were conducted with X-treme GENE HP DNA Transfection Reagent (Roche, Indianapolis, IN, USA) as manufacturer’s instruction.
IVs
IV strains A/PR8 (H1N1), A/duck/Hunan/3/2007 (H5N1) and A/Chicken/Jiangsu/7/2002 (H9N2) virus were propagated in the chorioallantoic fluid of 9-day-old embryonated specific-pathogen-free chicken eggs. LD50 was determined by intranasally inoculating adult mice, as described previously.27 For infection experiments, IVs were diluted in serum-free Dulbecco’s Modified Eagle’s Medium supplemented with 1% bovine serum albumin. A549 cells were infected at the indicated multiplicity of infection. After 1 h absorption at 37 °C, cells were washed with PBS and maintained in Dulbecco’s Modified Eagle’s Medium with 1% bovine serum albumin and 1 μg ml−1 TPCK-trypsin.
In vitro amiRNA screening
To screen amiRNAs for inhibition of IV replication, HEK 293 T (plated at 1 × 105 per well in 24-well plate on the previous day) were transfected with amiRNA expression plasmids. pcDNA 6.2-GW/miR empty vector served as a mock control. 24 h later, treated cells were infected with A/PR8 at 0.0005 multiplicity of infection. Cell lysates were collected to determine viral RNA copies by reverse transcription-PCR 24 h after virus infection.
Luciferase assay
A reporter plasmid driven by PolI promter was constructed as previously described.20 Firefly luciferase open reading frame was flanked by influenza NP noncoding regions. PB2, PB1, PA and NP genes of A/PR8 were cloned into expression plasmids pcDNA3.1(+) (Invitrogen, Grand Island, NY, USA). pGL4.74 (hRluc/TK) (Promega, San Luis Obispo, CA) continuously expressing Renilla luciferase was used as internal control. To evaluate the inhibition efficiency of NP-specific amiRNA on RNP activity, HEK 293 T cells were transfected with amiRNA expression plasmid, firefly luciferase reporter plasmid, expression plasmids of PR8-derived PB2, PB1, PA and NP (0.25 μg each) and pGL4.74 (0.1 μg). Twenty-four hours later, discard supernatant and lysed cells for dual-luciferase assays. Luciferase activity was measured by a Victor3 luminometer (PerkinElmer, Waltham, MA, USA) and normalized to Renilla activity accordingly. Experiments were repeated independently for three times.
Construction and production of Ad vectors
A chimpanzee origin Ad vector named AdC68 was chosen as a delivery vector. Ad vectors expressing amiRNA or chained amiRNAs were generated as follows:28 the complete expression cassettes of the miRNA(s) including the cytomegalovirus promoter and the TK pA were amplified from the pcDNA 6.2-GW/miR vectors by PCR and cloned into the pShuttle vector (Clonetech, Mountain View, CA, USA). Upon digestion with I-CeuI and PI-SceI, the amiRNA expression cassette was inserted into the E1 domain of AdC68 molecular clone. Recombinant Ad vectors were rescued by transfection of PacI linearized plasmid DNAs into HEK 293 cells. Ad vectors were amplified in HEK 293 cells and then purified by cesium chloride density-gradient centrifugation. Vp content was determined by spectrophotometry at 260 nm. By using same strategy, pcDNA 6.2-GW/miR empty vector was cloned into AdC68 vector. Recombinant virus, termed AdC68-miR-empty, was rescued and used as mock control virus in both in vitro and in vivo studies.
Western blot analysis
To verify that amiRNA(s) encoded by the Ad vectors reduced expression of the targeted proteins, NP and M2 protein was tested for by western blots in cells treated with the Ad vectors and then infected with A/PR8. In brief, A549 cells (plated at 1 × 105 per well in 24-well plate on the previous day) were inoculated with Ad-amiRNA or control Ads at the indicated doses. Cells treated with the 1010 vp of AdC68-miR-empty served as controls. Twenty-four hours later, cells were infected with A/PR8 virus at a dose of 0.001 multiplicity of infection. Twenty-four hours later, cells were harvested and western blotting was performed as previously described.29 To evaluate the structural protein expression over time, 1010 vp of Ads treated cells were collected 12 h, 24 h and 36 h post influenza infection. Protein expression levels were detected with primary antibodies for NP or M2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by secondary antibodies conjugated with horseradish peroxidase (Sigma–Aldrich, St Louis, MO, USA). The expression of β-actin (Sigma–Aldrich) was measured as a normalization control for protein loading. Signals were detected using a chemiluminescence detection system (GE Health Life Sciences, Pittsburgh, PA, USA).
Mice challenge experiments
Female ICR and C57Bl/6 mice aged 6–8 weeks were purchased from Shanghai laboratory animal center, China and housed in the biosafety level 2 facilities at Institute Pasteur of Shanghai. H5N1 challenge experiments were conducted in biosafety level 3 facilities at Fudan University. All animal experiments were conducted with protocols approved by Institutional Animal Care and Use Committee of Institute Pasteur of Shanghai. Groups of female ICR mice were inoculated intranasally with 1 × 1011 vp of Ad-amiRNAs. The same numbers of mice were inoculated with 1 × 1011 vp AdC68-miR-empty to control each experiment. 1, 5, 7, 10, 15 and 30 days later, mice were challenged intranasally with five LD50 of influenza A/PR8, H5N1 or H9N2, respectively, given in a volume of 30 μl. Body weights and survival rates were monitored daily for up to 21 days post infection. Mice, which lost over 30% of their initial weight, were killed for humanitarian reasons. For study of virus titer and histological changes in the mouse lungs, five mice in each group were euthanized and dissected 5 days post infection. All animal experiments were repeated at least three times in ICR mice.
Tetramer staining and intracellular cytokine staining
Lymphocytes were isolated from peripheral blood of C57Bl/6 mice 14 or 28 days post infection and stained with MHC I H-2Db NP peptide tetramer (ASNENTETM, Tetramer Core Facility, Emory University, GA, USA) and anti-CD8a-PerCP-Cy5.5 antibody (BD Biosciences, San Jose, CA) at 4 °C for 45 min. Flow cytometry was performed on BD LSR2 (BD Biosciences) and fluorescence-activated cell sorting data were analyzed by FlowJo software (TreeStar, Ashland, OR, USA).
Virus specific CD8+ T-cell responses were evaluated by staining for IFN-γ as previously described.30 Isolated lymphocytes (2 × 106 cells) were stimulated with IV NP peptide (2 μg ml−1), and stained with anti-CD8a-PerCP-Cy5.5 antibody. Samples were fixed, permeabilized and stained with fluorescein isothiocyanate-conjugated anti-IFN-γ (BD Pharmingen, San Diego, CA, USA). Samples were analyzed as described above.
Viral load measurement
Viral RNA copy numbers were determined by the previously validated reverse transcription-PCR method, which was modified to increase sensitivity.27, 31 Cells were collected for RNA extraction by TRIzol reagent according to manufacturer’s instruction (Invitrogen). The RNAs were diluted in 25 μl of diethylpyrocarbonate-treated water (Takara, Dalian, China) and RNA concentration was determined spectrophotometrically at an absorbance of 260 nm. From the entire RNA solutions, 100 ng RNA was applied for complementary DNA synthesis in 25 μl reaction volumes with the manufacturer-specified component proportions of a M-MLV Reverse Transcriptase Kit (Promega). Viral complementary DNA was quantified on 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA) by SYBR Premix Ex Taq kit (Takara) with following primers: M forward 5′-AAGACCAATCCTGTCACCTCTGA-3′, M reverse 5′-CAAAGCGTCTACGCTGCAGTCC-3′. The M genes of A/PR8 were cloned into the pMD18-T vector to provide a standard curve by 10-fold serial dilution. The reaction totaled 20 μl, containing 10 μl SYBR Premix Ex Taq (Tli RNaseH Plus) (2 × ), 0.4 μl PCR forward primer (10 μM), 0.4 μl PCR reverse primer (10 μM), 0.4 μl ROX reference dye2 (50 × ), 2 μl complementary DNA and 6.8 μl ddH2O. Real-time PCR was performed as follows: initially denaturation at 95 °C for 30 s, then 40 times cycling at 95 °C for 5 s, and 60 °C for 30 s. All samples were amplified in triplicate for each reaction. The data were analyzed on 7900HT System SDS Version 2.4 (Applied Biosystems).
Viral RNA copies in lung tissue were determined 5 days post infection as previously described.21 Lungs from dissected mice were homogenized in ice-cold PBS and centrifuged at 12000 rpm for 10 min. Supernatant of tissue homogenate were collected for viral titration as described above. Viral copy numbers were normalized to the mass of original tissues.
Histology
The lungs from dissected mice 5 days post infection were submerged in 10% formalin for tissue fixation for 24 h at 4 °C. They were stained with hematoxylin and eosin as described previously.27 Histopathological changes between different groups were compared in a double-blinded fashion to ensure objecitive evaluations. Lung pathology was scored as follows:27 (1) no observable pathology; (2) perivascular infiltrates; (3) perivascular and interstitial infiltrates affecting <20% of the lobe section; (4) perivascular and interstitial infiltrates affecting 20–50% of the lobe section; (5) perivascular and interstitial infiltrates affecting >50% of the lobe section.
Statistical analyses
Statistical analyses were performed with SPSS 16.0 (Chicago, IL, USA). One-way analysis of variance was applied to compare the relative messenger RNA levels, viral RNA copies, pathology grade and immune responses between groups. χ2-test was performed to compare the survival rates. P<0.05 was considered statistically significant.
References
Thompson WW, Comanor L, Shay DK . Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J Infect Dis 2006; 194 (Suppl 2): S82–S91.
Osterholm MT, Kelley NS . Mammalian-transmissible H5N1 influenza: facts and perspective. MBio 2012; 3: e00045–12.
Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 2013; 368: 1888–1897.
Gao GF . Influenza and the live poultry trade. Science 2014; 344: 235.
Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet 2014; 383: 714–721.
Qi W, Zhou X, Shi W, Huang L, Xia W, Liu D et al. Genesis of the novel human-infecting influenza A(H10N8) virus and potential genetic diversity of the virus in poultry, China. Euro Surveill 2014; 19.
Pica N, Palese P . Toward a universal influenza virus vaccine: prospects and challenges. Annu Rev Med 2013; 64: 189–202.
Tompkins SM, Lo CY, Tumpey TM, Epstein SL . Protection against lethal influenza virus challenge by RNA interference in vivo. Proc Natl Acad Sci USA 2004; 101: 8682–8686.
Hannon GJ . RNA interference. Nature 2002; 418: 244–251.
Jacque JM, Triques K, Stevenson M . Modulation of HIV-1 replication by RNA interference. Nature 2002; 418: 435–438.
Kapadia SB, Brideau-Andersen A, Chisari FV . Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc Natl Acad Sci USA 2003; 100: 2014–2018.
McCown M, Diamond MS, Pekosz A . The utility of siRNA transcripts produced by RNA polymerase i in down regulating viral gene expression and replication of negative- and positive-strand RNA viruses. Virology 2003; 313: 514–524.
Ge Q, Filip L, Bai A, Nguyen T, Eisen HN, Chen J . Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc Natl Acad Sci USA 2004; 101: 8676–8681.
Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN et al. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci USA 2003; 100: 2718–2723.
Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res 2007; 35: 5154–5164.
Bhomia M, Sharma A, Gayen M, Gupta P, Maheshwari RK . Artificial microRNAs can effectively inhibit replication of Venezuelan equine encephalitis virus. Antiviral Res 2013; 100: 429–434.
Ibrisimovic M, Kneidinger D, Lion T, Klein R . An adenoviral vector-based expression and delivery system for the inhibition of wild-type adenovirus replication by artificial microRNAs. Antiviral Res 2013; 97: 10–23.
Xie PW, Xie Y, Zhang XJ, Huang H, He LN, Wang XJ et al. Inhibition of Dengue virus 2 replication by artificial micrornas targeting the conserved regions. Nucleic Acid Ther 2013; 23: 244–252.
Lafforgue G, Martinez F, Sardanyes J, de la Iglesia F, Niu QW, Lin SS et al. Tempo and mode of plant RNA virus escape from RNA interference-mediated resistance. J Virol 2011; 85: 9686–9695.
Zhu W, Zhou J, Qin K, Du N, Liu L, Yu Z et al. A reporter system for assaying influenza virus RNP functionality based on secreted Gaussia luciferase activity. Virol J 2011; 8: 29.
Kim EH, Park HJ, Han GY, Song MK, Pereboev A, Hong JS et al. Intranasal adenovirus vectored vaccine for induction of long-lasting humoral immunity-mediated broad protection against influenza in mice. J Virol 2014; 88: 9693–9703.
Tatsis N, Ertl HCJ . Adenoviruses as vaccine vectors. Mol Ther 2004; 10: 616–629.
Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D et al. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood 2007; 110: 1916–1923.
Fan J, Liang X, Horton MS, Perry HC, Citron MP, Heidecker GJ et al. Preclinical study of influenza virus A M2 peptide conjugate vaccines in mice, ferrets, and rhesus monkeys. Vaccine 2004; 22: 2993–3003.
Berthoud TK, Hamill M, Lillie PJ, Hwenda L, Collins KA, Ewer KJ et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin Infect Dis 2011; 52: 1–7.
Lewis DL, Hagstrom JE, Loomis AG, Wolff JA, Herweijer H . Efficient delivery of siRNA for inhibition of gene expression in postnatal mice. Nat Genet 2002; 32: 107–108.
Zhou D, Wu TL, Lasaro MO, Latimer BP, Parzych EM, Bian A et al. A universal influenza A vaccine based on adenovirus expressing matrix-2 ectodomain and nucleoprotein protects mice from lethal challenge. Mol Ther 2010; 18: 2182–2189.
Zhou D, Zhou X, Bian A, Li H, Chen H, Small JC et al. An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nat Protoc 2010; 5: 1775–1785.
Wang S, Chi X, Wei H, Chen Y, Chen Z, Huang S et al. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J Virol 2014; 88: 8375–8385.
Fitzgerald JC, Gao GP, Reyes-Sandoval A, Pavlakis GN, Xiang ZQ, Wlazlo AP et al. A simian replication-defective adenoviral recombinant vaccine to HIV-1 gag. J Immunol 2003; 170: 1416–1422.
Youil R, Su Q, Toner TJ, Szymkowiak C, Kwan WS, Rubin B et al. Comparative study of influenza virus replication in Vero and MDCK cell lines. J Virol Methods 2004; 120: 23–31.
Acknowledgements
This work was supported by grants from Natural Science Foundation of China (31170871, 31370929 and 31200698), ‘Knowledge Innovation Program’ and ‘100 Talent Program’ from Chinese Academy of Sciences, the Shanghai Pasteur Foundation, China 863 program (2014AA021003). Partial support was also provided by Postdoctoral Science Foundation of China (2013T60470 and 2013M541559) and Postdoctoral Science Foundation of Shanghai Municipality, China (12R21416900). We gratefully acknowledge Jiawei Wang, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, for his generous assistance and reagents.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, H., Tang, X., Zhu, C. et al. Adenovirus-mediated artificial MicroRNAs targeting matrix or nucleoprotein genes protect mice against lethal influenza virus challenge. Gene Ther 22, 653–662 (2015). https://doi.org/10.1038/gt.2015.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2015.31
This article is cited by
-
Hemagglutinin-targeting Artificial MicroRNAs Expressed by Adenovirus Protect Mice From Different Clades of H5N1 Infection
Molecular Therapy - Nucleic Acids (2016)
