
Abstract
Dendritic cell (DC)-based vaccine approaches are being actively evaluated for developing immunotherapeutic agents against cancers. In this study, we investigated the use of engineered DCs expressing transgenic tumor-associated antigen hgp100 and the regulatory cytokine interleukin-21, namely DC-hgp100/mIL-21, as a therapeutic vaccine against melanoma. Tumor-bearing mice were injected intratumorally with transgenic DCs followed by three booster injections. Transgenic DC-hgp100/mIL-21 showed significant reduction in primary tumor growth and metastasis compared with DC-hgp100 alone and DC-mIL-21 alone. In vivo depletion of specific immune cell types (CD8+ T, CD4+ T and Natural killer (NK)-1.1+ cells) effectively blocked the protective effect of this combinational vaccine. In adoptive transfer experiments, a survival rate of nearly 90% was observed at 60 days post-tumor inoculation for the combinational vaccine group. In contrast, all mice in the DC-hgp100 and DC-mIL-21-only groups died within 43–46 days after tumor challenge. Considerably increased levels of interferon (IFN)-γ, tumor necrosis factor (TNF)-α, granulocyte macrophage colony-stimulating factor (GM-CSF) and cytotoxic T lymphocytes (CTLs) were detected with the combination vaccine group compared with other individual treatment groups. In comparison with the DC-hgp100 or mIL-21 groups, the combinational DC-hgp100/mIL-21 vaccine also drastically suppressed the myeloid-derived suppressor cells (MDSCs) and T-regulatory (Treg) cell populations. Our findings suggest that a combinational DC- and gene-based hgp100 and mIL-21 vaccine therapy strategy warrants further evaluation as a clinically relevant cancer vaccine approach for human melanoma patients.
Similar content being viewed by others


Introduction
Dendritic cells (DCs) are potent antigen-presenting cells (APCs)1 that have been extensively employed to initiate or enhance the presentation of tumor-associated antigens (TAA) or augment cytokine/chemokine-specific immune responses in animal models and clinical trials. A number of studies have shown that DCs modified ex vivo by pulsing with tumor lysates or TAA peptides can induce enhanced antitumor immunity.2, 3, 4, 5 Recently, the FDA approved a new prostate cancer vaccine, called Provenge (Dendreon), which uses patient’s own DCs for therapeutic vaccination.6 Specific virus-based vector systems hand have been reported to confer a high DC transduction efficiency and are able to generate strong and sustained CD4+ T-helper cell activity and cytotoxic T-cell responses against test tumors.4,7, 8, 9, 10, 11, 12 Recombinant modified vaccinia virus Ankara (rMVA) vectors have been shown to initiate a cascade of viral gene expression, allowing for the engineering of recombinant transgenes under the transcriptional control of vaccinia virus late promoters, which result in abundant synthesis of heterologous transgenic proteins—for example, various TAAs. DCs have been used with rMVA as vaccines to express transgenic antigens and cytokine adjuvants designed to induce a highly potent, target antigen-specific, immune context of DC-mediated signaling systems.13,14 Findings on significant activation of human gp100 and tyrosinase-specific CD8+ T cells have been reported, suggesting an efficient transgenic antigen presentation upon rMVA infection of DCs.13,15,16
We have previously reported that human gp100 (hgp100) is a promising target antigen for melanoma, which is capable of inducing specific humoral and cellular-mediated immunity.13,17 Immunization with hgp100-expressing viral vectors was shown to overcome self-tolerance to mouse gp100 in C57BL/6 mice because the approach is able to induce hgp100-specific as well as murine B16 melanoma-cross-reactive T-cell responses.18, 19, 20, 21 Interleukin-21 (IL-21) is a type I cytokine that shares a common cytokine receptor γ chain with other members of the IL-2 family. IL-21 can act as a key element in driving the transition from NK cell responses to specific cytotoxic T lymphocyte (CTL) responses. In T cells, IL-21 can block IL-2-induced apoptosis and promote the differentiation and long-term survival of CD8+ T cells.22 Tumor cells genetically engineered to secrete IL-21 were shown to induce protective immunity, abrogate T-regulatory (Treg) cells, recruit NK and CD8+ T cells and mediate expression of interferon (IFN)-γ.23 We have reported that granulocyte macrophage colony-stimulating factor (GM-CSF) complementary DNA (cDNA) and chemotactic chemokine CCL5 (RANTES) can enhance antitumor immunity induced by hgp100 DNA vaccination in a mouse melanoma model.13,24 However, as most tumors are known for not being rich in DCs, we consider that one therapeutic strategy is to take advantage of an engineered tumor microenvironment and to inject or target an abundant number of autologous DCs directly into the tumor tissues.
With the above considerations, we hence evaluated whether an overexpression of transgenic IL-21 in DCs can significantly enhance the efficacy of an engineered hgp100-DC vaccine on melanoma and whether such induction of antitumor immunity could be mediated via the inhibition of relevant immunosuppressive cells (for example, myeloid-derived suppressor cells (MDSCs) and Treg cells).7,8,25 Our finding from this study shows that the combinational use of DCs and rMVA vectors to deliver transgenically specific TAA and cytokine adjuvants into the tumor microenvironment may provide a useful approach as cell- and gene-based cancer vaccines. Specifics on possible future application of DC-based cancer vaccines against melanoma in cancer patients are discussed.
Results
Transgene expression in mature transduced DCs
For the appropriate pursuit of the designed gene and cell-based vaccine approach, we first quantified the transgene expression of hgp100 and mIL-21 mRNAs in transduced DCs using real-time PCR analysis. A significant level of hgp100 transcripts was observed in DCs transduced with hgp100 cDNA or with a combination of hgp100/mIL-21 cDNAs (P<0.001). No hgp100 mRNA was detected in other tested treatments, which hence also showed that, in our test, there was no cross-reaction in this assay (Figure 1a). Expression of mIL-21 mRNA was significantly higher in the DC-mIL-21 and the combination of DC-hgp100/mIL-21 mRNA transgene constructs, as compared with other treatments (P<0.001). Constitutive expression of mIL-21 in DCs was found to be significantly higher in DC-hgp100 and DC-MVA vector (P<0.01) treatments compared with non-transduced DC only treatment (Figure 1b).

Quantification of transgene expression in rMVA-transduced mature DCs. Transgenic mRNA levels of hgp100 (a) and mIL-21 (b) in rMVA-transduced DCs. ***P<0.001, compared with DC-MVA vector; **P<0.01 compared with DC-only. N.D: not detectable.
Antitumor immunity by DC vaccination
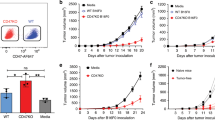
To investigate whether DC-based mIL-21 and DC-hgp100 transgene combination treatment enhances antitumor activity synergistically, we evaluated the efficacy of different DC-based vaccines in a B16/hgp100 melanoma mouse model. Vaccination with a combination of DC-hgp100/mIL-21 resulted in significantly higher reduction in tumor growth than vaccination with DC-hgp100 (95% confidence interval (CI)=163.9–459.3 mm3; P<0.001) on day 18 after tumor inoculation. On day 21, suppression of tumor growth under DC-hgp100/mIL-21 combination treatment was significantly increased compared with both DC-hgp100 (95% CI=293.8–571.6 mm3; P<0.001) and DC-mIL21 (95% CI=324.6–809.7 mm3; P<0.001) treatments. During this time period, the DC-MVA vector and DC only treatment had already resulted in tumors with sizes of >2000 mm3 (Figures 2a and b).

Protective immunity of rMVA-transduced DC vaccine against primary and metastatic melanoma. (a) Primary tumor model. C57BL/6 mice were inoculated with B16/hgp100 and vaccinated with rMVA-transduced DCs as described in Materials and methods. Data represent the means with upper 95% confidence intervals (n=8 mice per group) ***P<0.001, DC-hgp100/mIL-21 compared with the DC-hgp100 at 18 and 21 days after tumor inoculation. (b) Representative images of tumor appearance in mice receiving different treatments, as (i) PBS, (ii) DC only, (iii) DC-MVA vector, (iv) DC-hgp100, (v) DC-mIL-21 and (vi) DC-hgp100/mIL-21. (c) Survival time. **P<0.01, DC-hgp100/mIL-21 compared with the DC-hgp100 on day 46 post-tumor inoculation. (d) Metastasis model. C57BL/6 mice (n=8 each) in test groups were injected intravenously with B16/hgp100 cells and tumor metastasis into the lung was scored by estimating the percentage of surface area covered by metastasis as follows: 0, no metastasis; 1,<25%; 2, 25–50%; 3, 50–75%; and 4, >75% metastasis. Bars represent the mean metastasis scores for each treatment. *P<0.05 compared with DC-hgp100. (e) Assessment of immune cell subsets responsible for the DC-based vaccine immunity against melanoma. Depletion of CD4+ T7 CD8+ T cells and NK1.1+ cells was monitored using flow cytometry ***P<0.001 compared with anti-CD4+ and anti-NK-1.1+ cells. Experiments were repeated at least twice.
Survival time of mice vaccinated with DC-hgp100/mIL-21 was significantly longer than those vaccinated with DC-hgp100 and DC-mIL-21 (P<0.01) individually; of particular note, 80% of the mice treated with DC-hgp100/mIL-21 were still alive on day 46 post-tumor inoculation, as shown by log-rank test (Figure 2c). By contrast, all mice treated with DC-MVA vector and DC only died within 21 days and all mice treated with mIL-21 died within 46 days after tumor inoculation.
Next, the anti-metastatic effect of the DC-hgp100/mIL-21 combination vaccine was evaluated using a melanoma metastasis model. Mice were intravenously injected with B16/hgp100 tumor cells and vaccinated 5 days later. As shown in Figure 2d, tumor metastasis was significantly suppressed in the lung in response to DC-hgp100/mIL-21 combination treatment on 21 days post-tumor inoculation in comparison with other treatments (P<0.05).
Role of immune cell subsets in mediating tumor protection
To evaluate the importance of CD4+, CD8+ and NK-1.1+ cells in determining tumor protection, we injected anti-CD4+, anti-CD8+ or anti-NK-1.1+ monoclonal antibodies intraperitoneally before and after tumor inoculation and vaccination with DC-hgp100/mIL-21. All three immune cell subsets are apparently required for tumor protection (Figure 2e). Depletion of these immune cell subsets, most significantly CD8+ T cells, followed by CD4+ T cells and NK-1.1+ cells, blocked the protective effect of this combinational DC-hgp100/mIL-21 vaccine therapy. The rat IgG group treated with DC-hgp100/mIL-21 had a survival rate >80%, even on 60 days post-tumor inoculation (P<0.001 versus immune cell-depleted groups).
Adoptive transfer immune response in tumor protection
To further evaluate the role of combination vaccine therapy in tumor protection, splenocytes from B16/hgp100-inoculated, DC-hgp100/mIL-21 vaccine-treated tumor-free mice as well as splenocytes from other treatment and control groups were transferred intravenously to naive mice. One day later, the mice were challenged with B16/hgp100 tumor cells. Mice that received splenocytes from the combination DC-hgp100/mIL-21 vaccine-treated mice, but not from other test mice, were highly protected against a subsequent tumor challenge with B16/hgp100 cells and had greatly increased survival rate (P<0.001) compared with other groups (Figure 3a). Nearly 90% of these mice were alive on day 60 post-tumor inoculation, whereas all mice in the DC-hgp100 and DC-mIL-21 groups died within 43–46 days after tumor challenge. No tumor protection was observed in mice treated with PBS, DC-only and DC-MVA vector, mirroring the results from the therapeutic vaccination experiment (Figure 2c).

Adoptive transfer immune responses and induction of CD8+ T-cell-based immune responses of DC-based vaccine against melanoma. (a) Transfer of splenocytes from rMVA-transduced DC-vaccinated mice to naive mice were performed as described in Materials and methods. ***P<0.001, as DC-hgp100/mIL-21 group compared with DC-mIL-21 on day 60 after tumor inoculation. (b) IFN-γ-secreting CD8+ T-cell levels. CD8+ T cells from splenocytes were collected from test mice, stimulated and quantified ex vivo using ELISPOT assay. Results are expressed as IFN-γ-positive spot-forming CD8+ T cells (SFCs; 105). Data are the means of three independent experiments. ***P<0.001 compared with DC-hgp100; **P<0.01 compared with DC-MVA vector. (c) B16/hgp100 tumor-specific CTL activity in vaccinated and control mice. CD8+ T cells obtained from vaccinated or control mice were tested against target cells at the indicated effector:target ratios, and results are expressed as specific lysis (%). Data are the means of three independent experiments. ***P<0.001, **P<0.01 compared with indicated effector:target ratio of DC-hgp100.
Immune responses are enhanced by combination vaccine therapy
To analyze and compare the enhancement of immune responses by individual versus combination DC-based vaccinations, we collected CD8+ T cells from vaccinated and control mice at particular time points after treatments. Enzyme-linked immunospot (ELISPOT) assay showed much higher numbers of IFN-γ-secreting cells in the DC-hgp100/mIL-21 treatment group compared with the DC-hgp100 (95% CI=221–309.7; P<0.001) and DC-mIL-21 (95% CI=259.7–333.6; P<0.001) groups (Figure 3b). In turn, the individual treatment DC-hgp100 (95% CI=153.4–231.2; P<0.01) and DC-mIL-21 (95% CI=130.9–191.1; P<0.01) groups contained more IFN-γ-secreting cells than the DC-MVA vector group. These results suggest that the combination of hgp100 and mIL-21 apparently has a synergistic activity that can confer an improved protection against B16/hgp100 tumor in mice.
Next, we evaluated the specific cytolytic activity of CD8+ T cells induced by vaccination of mice with the various treatments. Cytotoxic CD8+ T cells from mice vaccinated with the DC-hgp100/mIL-21 combination had a significantly higher cytotoxic effect, killing more B16/hgp100 tumor cells than CD8+ T cells from the DC-hgp100 treatment group at effector-to-target cell ratios of 100:1 (95% CI=22.67–28.77%; P<0.001), 50:1 (95% CI=14.83–18.89%; P<0.001), 25:1 (95% CI=8.88–12.53%; P<0.001) and 12.5:1 (95% CI=3.28–5.23%; P<0.01; Figure 3c). Similar results were observed for the DC-mIL-21 versus the combinational vaccine group. Furthermore, a substantially higher cytotoxic effect was observed in the DC-hgp100 and DC-mIL-21 groups in comparison with the DC-MVA vector, DC only or PBS groups (Figure 3c).
To further characterize these immune responses, we used ELISA to determine the release of cytokines IFN-γ, IL-10, tumor necrosis factor (TNF)-α and GM-CSF from splenocytes 48 h after stimulation with MMC-treated B16/hgp100 cells. Secretion of the Th1 cytokine IFN-γ from splenocytes was two- to threefold higher in the DC-hgp100/mIL-21 combination treatment group than in the DC-hgp100 (95% CI=1220–1362 pg ml−1; P<0.001) and DC-mIL-21 (95% CI=1469–1596 pg ml−1; P<0.001) groups (Figure 4a). Interestingly, we also found that secretion of the Th2 cytokine IL-10 from splenocytes of the DC-hgp100/mIL-21 combination treatment group was much lower than in the DC-MVA vector or PBS control groups; it was also significantly lower than in the individual DC-hgp100 (95% CI=29.11–82.46 pg ml−1; P<0.01) and DC-mIL-21 (95% CI=47.51–92.56 pg ml−1; P<0.01) treatment groups (Figure 4b). There was no statistically significant difference in IL-10 secretion between the DC-hgp100 and DC-mIL-21 groups (95% CI=17.91–46.41 pg ml−1; P<0.286). We also determined the expression of two other important cytokines, TNF-α and GM-CSF. TNF-α secretion in the DC-hgp100/mIL-21 treatment group was considerably higher than in the DC-hgp100 (95% CI=159.5–238.8 pg ml−1; P<0.001) and DC-mIL-21 (95% CI=194.5–275.4 pg ml−1; P<0.001) groups (Figure 4c); similarly, GM-CSF secretion was also increased in the DC-hgp100/mIL-21 group as compared with the DC-hgp100 (95% CI=125.3–229.6 pg ml−1; P<0.01) and DC-mIL-21 (95% CI=156.9–265.3 pg ml−1; P<0.01) groups (Figure 4c).

Vaccination using rMVA-transduced-DCs expressing transgenic hgp100 and mIL-21 offers antigen-specific Th1 immunity. Splenocytes from test mice (n=3) were cocultured with MMC-treated B16/hgp100 tumor cells for 48 h, and supernatants from triplicate cultures/mouse were analyzed for IFN-γ (a), IL-10 (b), TNF-α and GM-CSF (c) by ELISA. ***P<0.001, **P<0.01 compared with DC-hgp100. (d) Expression of cytokine IFN-γ, IL-10, IL-17 and transcription factor T-bet-positive CD4+ T cell in vaccine-immunized mice. Seven days after last vaccination, spleen cells were collected from immunized mice, stimulated with phorbol 12-myristate 13-acetate and ionomycin, and labeled with anti-CD4-FITC, and stained for intracellular detection of IFN-γ, IL-10, IL-17 and T-bet. The percentage of cytokine-positive, CD4+ T cells is shown.
As T-bet is a critical transcription factor for Th1 cell differentiation26 and IFN-γ is a Th1 cytokine that also has an important role in cell-mediated immunity, we have further analyzed and compared the effect of DC-hgp100/mIL-21 combination vaccine on the expression of these functional molecules in CD4+ splenocytes using flow cytometry analysis. In this experiment, different subpopulations of CD4+ cells were compared for such molecular specificities. By comparison, we found that the populations of IFN-γ CD4+ T cells and T-bet+ CD4+ cells were drastically increased in DC-hgp100/mIL-21-vaccinated mice; however, this combinational vaccine also reduced the populations of IL-10+ CD4+ and IL17+ CD4+ cells in mice as compared with those of the PBS control and DC-MVA vector group mice (Figure 4d). In consistent with the result of IFN-γ secretion level (Figure 4a), these findings suggest that DC-hgp100/mIL-21 combination vaccine can effectively induce precursor CD4+ T cells toward the differentiation into Th1 cells, exhibiting an enhanced antigen presentation activity.
Combination DC vaccine therapy induces potent inhibition of immunosuppressive MDSC and Treg cells
The importance of Treg and MDSC in the regulation of tumor growth is now well documented. We hence went on to analyze the effect of various test vaccines on the inhibition of MDSC and Treg cells in immunized animals. There are two main subsets of MDSCs, the monocytic (CD11b+Ly6C+Ly6G-) and granulocytic (CD11b+Ly6ClowLy6G+) MDSC cells. The monocytic cells were previously shown to inhibit T-cell proliferation in vitro, whereas the granulocytic MDSCs were found to not inhibit such T-cell proliferation.27 In our present study, as shown in Figure 5a, the populations of monocytic MDSCs in spleen, blood, bone marrow and tumor infiltrating lymphocyte (TIL) tissues of various vaccinated mice were detected and compared. As compared with the DC-MVA vector group, the MDSC levels in DC-hgp100/mIL-21-vaccinated mice were significantly reduced to ~57% (26.1% versus 45.7%) in blood, and between 75 and 85% levels were detected for spleen, bone marrow and TIL tissues. For the single transgene-vaccinated groups (DC-mIL-21 and DC-hgp100), little or no effect was observed for most tested tissues, except for the TIL tissue samples, whereas a reduction to 53% was observed for DC-hgp100-vaccinated mice. On the other hand, the population of granulocytic MDSCs was drastically decreased to only 7% in TIL tissues of DC-hgp100/mIL-21-vaccinated mice; there was no significant change (that is, <10%) in the percentage of granulocytic MDSCs in spleen, blood or bone marrow tissues, as compared with those in DC-MVA vector-treated groups. These results suggest that the DC-hgp100/mIL-21 combination vaccine can most effectively suppress the monocytic MDSCs in blood tissues, whereas the level of granulocytic MDSCs was only suppressed in the TIL tissues.

Organ-specific reduction of MDSC population in mice immunized with DC-based vaccines. (a) Effect of vaccines on levels (%) of monocytic (M) and granulocytic (G) MDSCs in various tissues of test mice, quantified using flow cytometry. MDSC subpopulations were analyzed using the FACS DIVA software and gated on CD11b+ cells. (b) Suppression of Treg cell population in transgenic DC-vaccinated mice. Seven days post last vaccination, test cells were isolated from spleen, blood, bone marrow and TIL tissues of various DC vaccine- and control-immunized mice. The levels (%) of Treg cells were quantified using flow cytometry, as analyzed using the FACS DIVA software and gated on CD4+ cells.
The Treg cells, a subpopulation of the CD4+ T cells expressing CD25 and transcription factor FOXP3, are known to have a key role in promoting the growth and progression of tumors by inhibiting specific host immune response against the cancer.28,29 As shown in Figure 5b, we also compared the effect of different test vaccine groups on the population of Treg cells in different organs. We observed that the populations of Treg cells in bone marrow, spleen and TIL tissues were strongly suppressed in mice treated with the DC-hgp100/mIL-21 combination vaccine as compared with the DC-MVA vector-treated group. However, this suppressive effect on Treg cells was not detected in blood tissues tested. The single transgene vaccines, namely DC-mIL-21 and DC-hgp100, conferred some modest to substantial effect on bone marrow and TIL tissues but had little or no effect on spleen and blood tissues.
Inhibition of angiogenesis and increased immune cell infiltration into tumor tissues
Immunohistochemical staining analysis revealed that a large number of immune cells, mainly CD4+, CD8+ and NK-1.1 cell populations, were detected within test tumors in mice that were intratumorally injected with the DC-hgp100/mIL-21 combinational vaccine. In comparison, only a small number of immune cells infiltrated into test tumors in mice treated with PBS or DC-MVA vector (Figures 6a and b). In parallel, expression of hypoxia inducible factor-1α (HIF-1α), angiogenesis marker (vascular endothelial growth factor (VEGF)), platelet endothelial cell adhesion molecule (CD31) and matrix metalloproteinase-9 (MMP-9) was greatly suppressed in tumors of mice treated with the combinational vaccine as compared with that observed in tumors treated with PBS or DC-MVA vector (P<0.001).


Effect of DC-hgp100/mIL-21 vaccine on immune cell infiltration and suppression of angiogenic and MMPs cascade activities in treated mice, using immunohistochemical staining analysis. (a) Representative images of specific immune cells, including CD4+, CD8+ and NK-1.1+ cells, detected inside test tumors. (b) Quantification of the CD4+, CD8+, NK-1.1+ cell populations and the expression of HIF-1α, VEGF, CD31 and MMP-9-positive cells in test tumor tissues. Data were obtained as the means of the cell numbers counted from 10 different and representative microscopic fields ( × 200), with upper 95% confidence intervals from three mice per treatment group ***P<0.001 of DC-hgp100/mIL-21, as compared with PBS and DC-MVA vector groups.
Discussion
Suppression of tumor growth by host immune systems is now recognized as critical machinery for the control of progression versus regression of various human cancers. Many cancer immunologists believe that successful immunotherapy for cancer will likely be achieved by enhancing effector immune cells, whereas concomitantly inhibiting the immunosuppressive cells (for example, the MDSC and Treg cells involved in tumor tolerance) rather than by employing only one arm of these two approaches. Recently, DC-based cancer vaccines have attracted considerable attention as vehicles for the delivery of tumor-associated antigens, chemokines and cytokines.4,30 In this study, our DC-hgp100/mIL-21 combination vaccine strategy greatly enhanced the survival rate of test mice, especially during the late phase of tumor progression (Figure 1c). On the basis of our observations and knowledge, extension of lifespan of test mice to this extent is not common for most mouse tumor models. Similarly, compelling the same results were also observed in the therapeutic metastatic tumor model experiments (Figure 2d). All these enhanced in vivo antitumor effects suggest that a highly potent synergistic activity may be involved in the efficacy of the DC-hgp100/mIL-21 combinational vaccine in combating the growth of both primary and metastatic tumors.
Previous studies have reported that a DC-based hgp100 vaccine is effective against melanoma31 and that a DNA-based mIL-21 vaccine, either alone23,32 or in combination with other cytokines such as mIL-15,33 can exhibit significant antitumor activity against various cancer types. We observed in this study that IL-2134 and rMVA35 can confer the ability to suppress some DC maturation activities in response to stimuli such as LPS. However, the immunogenicity of rMVA/IL-21-transduced DC vaccine in our study is apparently dependent not only on the DC maturation activity but also on the expression of specific cytokines from test DCs and the molecular and cellular interaction between DC and other types of immune cells. On one hand, rMVA has been reported to serve as a very good adjuvant in cancer immunotherapy studies,16 and the DCs infected by rMVA can retain immunogenicity in vivo.7 On the other hand, IL-21 does have the ability to induce potent innate and adaptive immune responses.36 Additional studies showed that genetic modification of DCs and T cells with IL-21 could significantly enhance tumor-specific immunity.37,38 Furthermore, IL-21 not only could enhance proliferation, cytotoxicity and survival of CD8+ T cells39 but also can function as a pro-inflammatory cytokine, priming CD4+ T cells for differentiation into Th17 cells.40 In our current study, we found that IL-21 expression in DC vaccines could directly or indirectly suppress the populations of immunosuppressive MDSCs and Treg cells (Figure 5). These activities of DC vaccines may further modulate or enhance the immunity of CD8+ T cells, CD4+ T cells and NK cells, as shown in Figure 2e. Taken together, although the rMVA/IL-21-combined vaccine can partially suppress DC maturation as recently reported by Tao et al.,35 our results strongly suggest that this IL-21-enhanced tumor-specific immunity is highly potent, and it can overcome the associated suppressive effect on DC maturation. The observed vaccine effect, we consider, can be further induced by cytokine (IL-21)-derived tumor-specific immunity against tumors in vivo. We suggest that IL-21 may directly or indirectly enhance the tumor-specific immunity via the activation of CD8+ T cells, differentiation of CD4+ T cells and the activation of NK and NKT cells.
Whereas populations of IFN-γ+ CD4+ T cells and T-bet+ CD4+ cells, as Th1 cells, were drastically increased in DC-hgp100/mIL-21-vaccinated mice, reduced levels of IL-10+ CD4+ and IL-17+ CD4+ cells as Th2 cells were observed, as compared with control vector groups (Figure 4d). In consistency, enhanced levels for IFN-γ, TNF-α and GM-CSF secretion were also defected from splenocytes of these mice (Figures 4a–c). Our findings thus suggest mechanistically that the DC-hgp100/mIL-21 combination vaccine is mediated by Th1 but not by Th2 response. Our results may also imply that the high-level induction of IFN-γ, TNF-α, GM-CSF and CTLs in the combination group could activate DCs through an IFN-γ-dependent pathway and a subsequent enhancement of CTL activity as a specific cell-mediated immunity.
Inhibition of tumor-specific CD8+ effector T cells and elimination of MDSC and Treg in mouse tumor models have previously been shown to, result in efficacious tumor regression.4,23 In our present study, the population levels of monocytic MDSCs in spleen, blood, bone marrow and TIL tissues of DC-hgp100/mIL21-vaccinated mice were considerably lower than those in PBS or DC-MVA vector-vaccinated mice (Figure 5a). In contrast, there was no significant change in percentages of granulocytic MDSCs in spleen, blood and bone marrow as compared with those in PBS or empty vector-vaccinated mice. These results suggest that DC-hgp100/mIL-21 combination vaccine can preferentially suppress the population levels of monocytic MDSCs in various test organs; however, the granulocytic MDSCs were mainly, if not only, suppressed in the TIL tissues. The population levels of Treg cells in spleen, bone marrow and TIL tissues were found to be strongly suppressed by treatment with DC-hgp100/mIL21 combination vaccine as compared with PBS and DC-MVA vector-vaccinated group (Figure 5b). This suppressive effect on Treg cells, however, was not detected in blood tissues. The organismic and physiogical significance levels of these vaccine effects on MDSC and Treg cell populations are not clear to us based on the result from the present study. These findings, however, may indicate that a coordinated effect was involved in the inhibition of Treg cells in the tumor microenvironment, which could have resulted in a substantial reduction in tumor growth in vivo. As enhanced immune cell infiltration (mainly CD4+, CD8+ and NK-1.1+ cells) into the test tumor microenvironment and the concomitant suppression of VEGF, CD31, MMP-9 and HIF-1α activities were clearly detected for the combination DC-hgp100/mIL-21 vaccine therapy (Figures 6a and b), we consider that these activities in tumor may also curtail functional impairment of DCs in the tumor microenvironment that fails to induce a full antitumor immune response.
In conclusion, we have shown that a DC-hgp100/mIL-21 combination vaccine is able to effectively suppress primary and metastatic B16/hpg100 melanoma by enhancing various specific antitumor immune responses. This profound effect is likely mediated by timely immune cell proliferation and enhanced production of specific Th1 cytokines (IFN-γ) and a concomitant suppression of specific Th2 cytokines (IL-10), angiogenesis growth factor VEGF, platelet endothelial cell adhesion molecule CD31, HIF-1α, MMP-9 and the active expansion of effector CD4+ and CD8+ T cells and, in parallel, a potent inhibition of immunosuppressive MDSCs and Treg cells within the tumor microenvironment. In the future, this combination DC-hgp100/mIL-21 vaccine therapy approach needs to be further evaluated in preclinical and clinical studies for possible application to treatment of human melanoma patients.
Materials and methods
Animals
Female C57BL/6JNarl mice (6–8 weeks old) were purchased from the National Laboratory Animal Breeding and Research Center, Taipei, Taiwan. All mice were maintained in a laminar air flow cabinet in a room kept at 24±2 °C, with 40–70% humidity and a 12-h light/12-h dark cycle under specific pathogen-free conditions. All facilities were approved by the Academia Sinica Institutional Animal Care and Utilization Committee, and all animal experiments were conducted under the institutional guidelines established by the Animal Core Facility at Academia Sinica, Taipei.
Cell lines
The mouse B16 melanoma and hamster fibroblast BHK21 cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Tumor cell cultures were maintained in Dulbecco’s modified Eagle’s medium supplemented with 1.5 g l−1 sodium bicarbonate, 10% fetal bovine serum, 100 μg ml−1 streptomycin and penicillin, and 2 mM L-glutamine.
Recombinant MVA vector construction
For B16 cell transfection, a cDNA vector construct pNASS/CMV-hgp100 was generated by inserting an hgp100 cDNA fragment, excised from the pWRG1644 vector, into the pNASS/CMV-neo vector.41 B16 cells stably transfected with this hgp100 cDNA vector, designated as B16/hgp100, were obtained as previously reported.24 The hgp100 cDNA was amplified and subcloned into the SalI-PstI site of the pLW44 transfer vector (provided by Dr Bernard Moss, NIH, Bethesda, MD, USA), bringing it under the control of a vaccinia virus-modified H5 early–late promoter.42 The Igκ leader sequence was subcloned into the SmaI–SalI site of pLW44 for secretion of the transgene product, as previously described.13 Murine IL-21 cDNA was purchased from Invivogen (San Diego, CA, USA). Recombinant MVA was made by transfecting transfer plasmids into BHK-21 cells infected with 0.05 plaque-forming units of MVA per cell. Fluorescent plaques were cloned by eight successive rounds of plaque isolation and then propagated in BHK-21. Titers of rMVA were determined by staining the plaques with anti-vaccinia virus rabbit antibody.43
Preparation of bone marrow DCs
Bone marrow DCs were prepared as described previously with slight modifications.13,44 Briefly, Bone marrow DCs prepared from femur and tibia of C57BL/6JNarl mice were depleted of red blood cells with 0.84% Tris-ammonium chloride and plated in DC culture medium (RPMI plus 10% FBS, GM-CSF (20 ng ml−1) and IL-4 (20 ng ml−1)). Nonadherent granulocytes, T and B cells were gently removed and fresh medium was added on the fifth day. Nonadherent cells were harvested on the seventh day. DCs generated in this manner showed expression of CD11c, co-stimulatory factors (CD40, CD80 and CD86) and MHC class II in response to lipopolysaccharide for 24 h (data not shown). These well-differentiated mature DCs were then used for in vitro MVA DC transduction.
MVA transduction of bone marrow DCs
The rMVA-containing human gp100 and mIL-21 transgenes were constructed and DC transduction was performed as described previously with slight modification.45,46 In brief, DCs (2 × 106) were collected on the seventh day and resuspended in a six-well plate with 1 ml medium containing rMVA (with multiplicity of infection=5). The cells were then centrifuged, re-suspended with gentle shaking and rested for 1 h at 37 °C. The cells were washed again with medium, resuspended with 3 ml of conditioned medium containing lipopolysaccharide (1 μg ml−l) and then rested overnight in an incubator at 37 °C.
Mixed lymphocyte reaction
Mixed lymphocyte reaction was performed to evaluate the allostimulatory function of DCs as previously described.30 Immature and mature DCs transduced with MVA as described above were washed and cocultured with allogeneic (Balb/c) naive CD4 T cells (1 × 105 cells per well) in 96-well plates at indicated ratios. T-cell proliferation was measured on day 4 using a BrdU cell proliferation ELISA kit (Roche, Penzberg, Germany) according to the manufacturer’s instructions.
Real-time PCR
Quantification of hgp100 and mIL-21 mRNA expression from DCs obtained after various treatments was performed using high performance SYBR green real-time PCR (LightCycler, Roche, Indianapolis, IN, USA) as previously described.47 The primers contained the following sequences: human gp100 sense primer 5′-TATTGAAAGTGCCGAGATCC-3′ and antisense primer 5′-ACTGGTGCAGAACCAGCTG-3′; mouse IL-21 sense primer 5′-GCGTCGACATGGAGAGGACCCT-3′ and antisense primer 5′-GCTGCATGCTCACAGTGCCCCTTTA-3′; mouse GAPDH sense primer 5′-CATCACTGCCACCCAGAAGACTGTGGA-3′ and antisense primer 5′-TACTCCTTGGAGGCCATGTAGGCCATG-3′. Each measurement was repeated three times and GAPDH RNA was used as an internal control.
Animal experiments
Primary tumor model
C57BL/6 mice were divided into six experimental groups (eight mice per group). B16/hgp100 tumor cells were collected at 80% confluency, washed in phosphate-buffered saline (PBS), centrifuged (2500 g for 5 min), resuspended in Hanks’ balanced salt solution (Life Technologies, Rockville, MD, USA) and injected subcutaneously (105 cells per 100 μl per mouse) into the right flank of mice on day 0. One week after tumor inoculation (tumor size ~80–100 mm3), tumor-bearing mice were injected intratumorally with transgenic DCs (5 × 105 cell per 50 μl per mouse) expressing hgp100 and mIL21 followed by booster injections at 4-day intervals. The six treatments were as follows: (i) PBS, (ii) DCs only, (iii) DC-MVA vector, (iv) DC-hgp100, (v) DC-mIL-21 and (vi) DC-hgp100/mIL21. Test mice were examined twice weekly for tumor appearance and tumor volumes were determined from the length (a) and width (b) of test tumors as measured in a blinded manner by calipers using the formula: V=ab2/2. Survival time of mice was also observed.13
Metastasis model
C57BL/6 mice were divided into six experimental groups (eight mice per group). Each mouse, depending on the treatment, was injected intravenously with B16/hgp100 tumor cells (105 cells per 100 μl per mouse) on day 0, and 5 days later DC vaccines were delivered intravenously into test mice (therapeutic model). Vaccinated mice subsequently received two boosters at 4-day intervals. The experiment was terminated on day 21, and tumor metastasis into the lung was determined in a blinded manner. Each experiment was repeated at least twice.
Adoptive transfer of splenocytes
To determine whether lymphocytes induced by transgenic hgp100 and mIL21 could protect naive mice from a melanoma challenge, C57BL/6 mice were inoculated with B16/hgp100 tumor cells (105) and injected intratumorally with DC-hgp100, DC-mIL21 or DC-hgp100/mIL21 as described above. After 3 weeks, splenocytes were harvested and 2 × 107 cells were infused intravenously into naive mice. Control groups of mice received splenocytes from B16/hgp100 tumor-bearing mice treated with PBS, DCs only or DC-MVA vector. One day later, mice were subcutaneously challenged with B16/hgp100 (105) cells and monitored for tumor volume and survival.
In vivo depletion of immune cell subsets
Right flanks of C57BL/6 mice (n=7) were subcutaneously inoculated with B16/hgp100 tumor cells (105). One week later, tumor-bearing mice were injected intratumorally with transgenic DCs expressing hgp100 and mIL21 followed by two booster injections. Rat anti-CD4 (GK1.5), anti-CD8 (53-6.7) and anti-NK1.1 (PK136) monoclonal antibodies (100 μg per injection per mouse, all from BioLegend, San Diego, CA, USA) were used to deplete CD4+ T cells, CD8+ T cells and NK1.1+ cells, respectively, on days -3, 0, 5, 10, 15 and 20. Normal rat IgG was used as a negative control. Target cell depletion was monitored using flow cytometry of peripheral lymphocytes isolated from test mouse blood and stained with fluorescein isothiocyanate-conjugated anti-CD4+, CD8+ and anti-NK-1.1 (data not shown). Survival of mice was observed up to 60 days after tumor inoculation.13
IFN-γ enzyme-linked immunospot assay
Human gp100-specific IFN-γ-secreting CD8+ T cells from test mice were enumerated using enzyme-linked immunospot (ELISPOT) assay (R&D Systems, Minneapolis, MN, USA), as described previously with slight modifications.13 Briefly, nitrocellulose-backed plates (96-well, MAHA S45; Millipore, Bedford, MA, USA) were coated with murine IFN-γ-specific Ab R4 (BD PharMingen, San Jose, CA, USA) overnight at 4 °C. Test wells were washed five times with PBS and blocked using RPMI 1640 supplemented with 10% fetal bovine serum at 25 °C for 2 h. Freshly purified CD8+ T cells were isolated from the spleen (105 cells) by using mouse CD8 microbeads, and mitomycin C (MMC)-treated B16/hgp100 cells were then added into the wells and incubated for 24 h at 37 °C in 5% CO2. Purified CD8+ T cells without the MMC-treated B16/hgp100 cells were incubated as a negative control. IFN-γ spots were counted by using an immunospot analyzer and confirmed by a computer-based Immunospot software (Cellular Technology, Shaker Heights, OH, USA). Data from all the wells were averaged and normalized by comparing the ratio of antigen-specific spots to negative control spots.
Assay for CTL activity
To show that tumor-specific CTLs had been generated in the immunized or control mice (n=3), CD8+ T cells were collected 1 week after the booster vaccination and used as effector cells. Target cells (B16/hgp100) were Eu-labeled, and nonradioactive DELFIA EuTDA cytotoxicity assay was performed as described.13 Briefly, B16/hgp100 tumor cells were collected, washed once and labeled with DELFIA BATDA reagent (DELFIA, Wellesley, MA, USA) for 30 min at 37 °C. After three additional washes, 5 × 103 targets (per well) were incubated with effector CD8+ T cells at the indicated ratios of effector to target cells in 96-well, flat-bottom plates (DELFIA) for 2 h at 37 °C. The proportion of specific lysis was calculated as 100 × (experimental release (counts)-spontaneous release (counts))/(maximum release (counts)-spontaneous release (counts)).
ELISA for cytokine release
One week after administration of the second boost vaccine, splenocytes were collected from immunized or control mice (n=3) and washed three times with PBS. Splenocytes (2 × 106 cells per ml) were then stimulated with MMC-treated B16/hgp100 cells in a 10:1 ratio. Culture supernatants were collected after 48 h and assayed for the levels of IFN-γ, IL-10, TNF-α and GM-CSF using ELISA kits (R&D Systems).
Intracellular staining and flow cytometry assays
For intracellular staining of test cytokines, spleen cells were isolated from immunized mice and stimulated with 50 ng ml-l of phorbol 12-myristate 13-acetate (Sigma, St Louis, MO, USA) and 500 ng ml−1 of ionomycin for 6 h, and GolgiPlug (BD PharMingen, San Diego, CA, USA) was added during the last 2 h. Cells were then stained with fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD4 (Biolegend) for 30 min at 4 °C, subsequently stained with PE-Cy7-conjugated anti-mouse IFN-γ, APC-conjugated anti-mouse IL-10, Alexa Fluor 647-conjugated anti-mouse IL-17 and Per/CP-Cy5-conjugated anti-mouse T-bet, followed by cells permeabilized with the Cytofix/Cytoperm Plus kit (BD PharMingen) according to the manufacturer’s protocol. All antibodies and test kits were obtained from BD PharMingen. Fluorescence signals were detected by a BD LSRII flow cytometer and analyzed by a FACS DIVA software (BD Biosciences, San Jose, CA, USA). The percentage of various cytokine-expressing cells were gated on CD4+ T cells.
For preparation of MDSCs, spleen cells, bone marrow cells, peripheral blood cells and tumor infiltrating lymphocytes (TILs) from immunized and control mice were collected, and cells were stained for 30 min at 4 °C with antibodies against specific cell markers, including FITC-conjugated anti-mouse CD11b (for cell surface), APC-Cy7-conjugated anti-mouse Ly-6C and PE-conjugated anti-mouse Ly-6G (both for intracellular staining). All three antibodies were obtained from Biolegend. For Treg cell detection, cells were surface-stained with FITC-conjugated anti-mouse CD4, PE-conjugated anti-mouse CD25 and intercellularly stained with APC-conjugated anti-mouse Foxp3, followed by cell permeabilization with the Cytofix/Cytoperm Plus kit, according to the manufacturer’s protocol for all three antibodies and reagent kit from BD PharMingen. Fluorescence signals were detected using cytometry as described above. The percentages of MDSCs and Treg cells were gated on CD11b+ cell and CD4+ cell, respectively.48
Immunohistochemical staining analysis
After the indicated time periods, test tumors were harvested, snap-frozen and cut into 7-μm sections using a cryotome (Microm HM 550, Waltham, MA, USA). Before incubating with CD4, CD8, NK1.1, MMP-9, VEGF, CD31 or HIF-1α antibodies, the cryostat sections were placed on glass slides, air-dried and treated with 0.3% H2O2 for 10 min to block endogeneous peroxidase activities. Slides were sequentially incubated with diluted, specific primary antibodies overnight at 4 °C, and bound antibodies were detected using an anti-IgHRP detection kit (BD Biosciences).
Statistical analyses
Data are presented as the means with 95% CI of at least three experiments. Statistical analyses were carried out with GraphPad Prism 5.01 (San Diego, CA, USA). Groups were compared by Student’s t-test. Differences in survival time were evaluated by a log-rank test of the Kaplan–Meier survival curves. All statistical tests were two-sided. P-values of <0.05 were considered statistically significant.
References
Banchereau J, Steinman RM . Dendritic cells and the control of immunity. Nature 1998; 392: 245–252.
Banchereau J, Ueno H, Dhodapkar M, Connolly J, Finholt JP, Klechevsky E et al. Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother 2005; 28: 505–516.
Wang B, Zaidi N, He LZ, Zhang L, Kuroiwa JM, Keler T et al. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res 2012; 14: R39.
Huang J, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee M et al. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther 2009; 18: 264–274.
Lesterhuis W, de Vries IJ, Schuurhuis DH, Boullart AC, Jacobs JF, de Boer AJ et al. Vaccination of colorectal cancer patients with CEA-loaded dendritic cells: antigen-specific T cell responses in DTH skin tests. Ann Oncol 2006; 17: 974–980.
Brower V . Approval of provenge seen as first step for cancer treatment vaccines. J Natl Cancer Inst 2010; 102: 1108–1110.
Behboudi SMA, Gilbert SC, Nicoll CL, Hill AVS . Dendritic cells infected by recombinant modified vaccinia virus Ankara retain immunogenicity in vivo despite in vitro dysfunction. Vaccine 2004; 22: 4326–4331.
Kastenmuller W, Drexler I, Ludwig H, Erfle V, Peschel C, Bernhard H et al. Infection of human dendritic cells with recombinant vaccinia virus MVA reveals general persistence of viral early transcription but distinct maturation-dependent cytopathogenicity. Virology 2006; 350: 276–288.
Mossoba M, Walia JS, Rasaiah VI, Buxhoeveden N, Head R, Ying C et al. Tumor protection following vaccination with low doses of lentivirally transduced DCs expressing the self antigen erbB2. Mol Ther 2008; 16: 607–617.
Nabekura T, Otsu M, Nagasawa T, Nakauchi H, Onodera M . Potent vaccine therapy with dendritic cells genetically modified by the gene-silencing-resistant retroviral vector GCDNsap. Mol Ther 2006; 13: 301–309.
Van Tendeloo V, Snoeck HW, Lardon F, Vanham GLEE, Nijs G, Lenjou M et al. Nonviral transfection of distinct types of human dendritic cells: high-efficiency gene transfer by electrophoration into hematopoietic progenitor-but not monocyte-derived dendritic cells. Gene Ther 1998; 5: 700–707.
Yoshikawa T, Niwa T, Mizuguchi H, Okada N, Nakagawa S . Engineering of highly immunogenic long-lived DC vaccines by antiapoptotic protein gene transfer to enhance cancer vaccine potency. Gene Ther 2008; 15: 1321–1329.
Aravindaram K, Yu HH, Lan CW, Wang PH, Chen YH, Chen HM et al. Transgenic expression of human gp100 and RANTES at specific time points for suppression of melanoma. Gene Ther 2009; 16: 1329–1339.
Smith C, Dunbar PR, Mirza F, Palmowski MJ, Shepherd D, Gilbert SC et al. Recombinant modified vaccinia Ankara primes functionally activated CTL specific for a melanoma tumor antigen epitope in melanoma patients with a high risk of disease recurrence. Int J Cancer 2005; 113: 259–266.
Di Nicola M, Carlo-Stella C, Anichini A, Mortarini R, Guidetti A, Tragni G et al. Clinical protocol. Immunization of patients with malignant melanoma with autologous CD34(+) cell-derived dendritic cells transduced ex vivo with a recombinant replication-deficient vaccinia vector encoding the human tyrosinase gene: a phase I trial. Hum Gene Ther 2003; 14: 1347–1360.
Di Nicola M, Carlo-Stella C, Mortarini R, Baldassari P, Guidetti A, Gallino GF et al. Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD34 (+)-derived dendritic cell vaccination: a phase I trial in metastatic melanoma. Clin Cancer Res 2004; 10: 5381–5390.
Schuurhuis D, Verdijk P, Schreibelt G, Aarntzen EHJG, Scharenborg N, de Boer A et al. In situ expression of tumor antigens by mRNA electroporated dendritic cells in lymph nodes of melanoma patients. Cancer Res 2009; 69: 2927–2934.
Neal Z, Bates MK, Albertini MR, Herweijer H . Hydrodynamic limb vein delivery of a xenogeneic DNA cancer vaccine effectively induces antitumor immunity. Mol Ther 2007; 15: 422–430.
Perricone M, Claussen KA, Smith KA, Kaplan JM, Piraino S, Shankara S et al. Immuno gene therapy for murine melanoma using recombinant adenoviral vectors expressing melanoma associated antigens. Mol Ther 2000; 1: 275–284.
Wan Y, Emtage P, Zhu Q, Foley R, Pilon A, Roberts B et al. Enhanced immune response to the melanoma antigen gp100 using recombinant adenovirus-transduced dendritic cells. Cell Immunol 1999; 198: 131–138.
Zhai Y, Yang JC, Spiess P, Nishimura MI, Overwijk WW, Roberts B et al. Cloning and characterization of the genes encoding the murine homologues of the human melanoma antigens MART1 and gp100. J Immunother 1997; 20: 15–25.
Chaput ND-JG, Bergot A-S, Cordier C, Ngo-Abdalla S, Klatzmann D . Regulatory T cells prevent CD8 T cell maturation by inhibiting CD4 Th cells at tumor sites. J Immunol 2007; 179: 4969–4978.
Schulze S, Kim HS, Fan Q, Kim DW, Kaufman HL . Local IL-21 promotes the therapeutic activity of effector T cells by decreasing regulatory T cells within the tumor microenvironment. Mol Ther 2009; 17: 380–388.
Rakhmilevich A, Imboden M, Hao Z, Macklin MD, Roberts T, Wright KM et al. Effective particle-mediated vaccination against mouse melanoma by coadministration of plasmid DNA encoding gp100 and granulocyte-macrophage colony-stimulating factor. Clin Can Res 2001; 7: 952–961.
Trevor K, Hersh EM, Brailey J, Balloul JM, Acres B . Transduction of human dendritic cells with a recombinant modified vaccinia ankara virus encoding MUC1 and IL2. Cancer Immunol Immunother 2001; 50: 397–407.
Szabo S, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH . A novel transcription factor, T-bet directs Th1 lineage commitment. Cell 2000; 100: 655–669.
Gabrilovich DaN S . Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9: 162–174.
Shevach E . Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity 2009; 30: 636–645.
Yang Z, Zang B, Li D, Lv M, Huang C, Shen GX et al. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One 2010; 5: 1–9.
Tatsumi T, Gambotto A, Robbins PD, Storkus WJ . Interleukin 18 gene transfer expands the repertoire of antitumor Th1-type immunity elicited by dendritic cell-based vaccines in association with enhanced therapeutic efficacy. Cancer Res 2002; 62: 5853–5858.
Yang S, Vervaert CE, Burch J, Grichnik J, Seigler HF, Darrow TL . Murine dendritic cells transfected with human gp100 elicit both antigen specific CD8+ and CD4+ T-cell responses and are more effective than DNA vaccines at generating anti-tumor immunity. Int J Cancer 1999; 83: 532–540.
Li YaY C . IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood 2008; 111: 229–235.
Kowalczyk A, Wierzbicki A, Gil M, Bambach B, Kaneko Y, Rokita H . Induction of protective immune responses against NXS2 neuroblastoma challenge in mice by immunotherapy with GD2 mimotope vaccine and IL-15 and IL-21 gene delivery. Cancer Immunol Immunother 2007; 56: 1443–1458.
Brandt KB-PS, Foster DC, Rückert R . Interleukin-21 inhibits dendritic cell activation and maturation. Blood 2003; 102: 4090–4098.
Tao RLL, Huang W, Zheng L . Activation of human dendritic cells by recombinant modified vaccinia virus Ankara vectors encoding Survivin and IL-2 genes in vitro. Hum Gene Ther 2010; 98: 98–108.
Ma HL WM, Konz RF, Senices M, Young DA, Grusby MJ, Collins M et al. IL-21 activates both innate and adaptive immunity to generate potent antitumor responses that require perforin but are independent of IFN-γ. J Immunol 2003; 171: 608–615.
Kaka ASSD, Hartmaier R, Leen AM, Lu A, Bear A, Rooney CM et al. Genetic modification of T cells with IL-21 enhances antigen presentation and generation of central memory tumor-specific cytotoxic T-lymphocytes. J Immunother 2009; 32: 726–736.
Zeng RSR, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med 2005; 201: 139–148.
Ansén SBM, Berezovskaya A, Murray AP, Stevenson K, Nadler LM, Hirano N . Dissociation of its opposing immunologic effects is critical for the optimization of antitumor CD8+ T-cell responses induced by interleukin 21. Cancer Res 2008; 14: 6125–6136.
Spolski RLW . Cytokine mediators of Th17 function. Eur J Immunol 2009; 39: 658–661.
Albertini M, Emler CA, Schell K, Tans KJ, King DM, Sheehy MJ . Dual expression of human leukocyte antigen molecules and the B7–1 costimulatory molecule (CD80) on human melanoma cells after particle-mediated gene transfer. Cancer Gene Ther 1996; 3: 192–201.
Wyatt L, Shors ST, Murphy BR, Moss B . Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996; 14: 1451–1458.
Earl P, Moss B, Wyatt LS, Carroll MW . Generation of recombinant vaccinia viruses. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidmen ZG, Smith ZA, Skuhl LX (eds). Current Protocols in Molecular Biology. John Wiley and Sons: NewYork, NY, USA, 1998, pp 16.17.1–16.19.11.
Labeur M, Roters B, Pers B, Mehling A, Luger TA, Schwarz T et al. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol 1999; 162: 168–175.
Nagerson D, Panelli M, Dudley ME, Finkelstein SE, Rosenberg SA, Marincola FM . Biased epitope selection by recombinant vaccinia-virus (rVV)-infected mature or immature dendritic cells. Gene Ther 2003; 10: 1754–1765.
Yin SY, Wang CY, Yang NS . Interleukin-4 enhances trafficking and functional activities of GM-CSF-stimulated mouse myeloid-derived dendritic cells at late differentiation stage. Exp Cell Res 2011; 317: 2210–2221.
Loges S, Clausen H, Reichelt U, Bubenheim M, Erbersdobler A, Schurr P et al. Determination of microvessel density by quantitative real-time PCR in esophageal cancer: correlation with histologic methods, angiogenic growth factor expression, and lymph node metastasis. Clin Cancer Res 2007; 13: 76–80.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell–dependent antitumor immunity. Cancer Res 2010; 70: 3052–3061.
Acknowledgements
This work was supported by grants from the National Science Council (NSC 102-2320-B-001-002) and a grant (99-Academia Sinica investigator award-12) from the Academia Sinica, Taiwan. We thank Hsiu-Hui Yu, Yun-Hsiang Chen, Chun-Wen Lan, Li-Ting Huang and Yung-Tsung Chen for their technical help in carrying out this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Aravindaram, K., Wang, PH., Yin, SY. et al. Tumor-associated antigen/IL-21-transduced dendritic cell vaccines enhance immunity and inhibit immunosuppressive cells in metastatic melanoma. Gene Ther 21, 457–467 (2014). https://doi.org/10.1038/gt.2014.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2014.12
