
Abstract
This work illustrates the utility of Minivector DNA, a non-viral, supercoiled gene therapy vector incorporating short hairpin RNA from an H1 promoter. Minivector DNA is superior to both plasmid DNA and small interfering RNA (siRNA) in that it has improved biostability while maintaining high cell transfection efficiency and gene silencing capacity. Minivector DNAs were stable for over 48 h in human serum, as compared with only 0.5 and 2 h for siRNA and plasmid, respectively. Although all three nucleic acids exhibited similar transfection efficiencies in easily transfected adhesion fibroblasts cells, only Minivector DNAs and siRNA were capable of transfecting difficult-to-transfect suspension lymphoma cells. Minivector DNA and siRNA were capable of silencing the gene encoding anaplastic lymphoma kinase, a key pathogenic factor of human anaplastic large cell lymphoma, and this silencing caused inhibition of the lymphoma cells. Based on these results, Minivector DNAs are a promising new gene therapy tool.
Similar content being viewed by others


Introduction
Gene silencing for gene therapy remains challenging because of the limited biostability of small interfering RNA (siRNA) and low transfection efficiency of plasmid DNA. This paper describes the first cell transfection experiments with Minivector DNA, a novel, non-viral, supercoiled gene therapy vector as small as ∼300 bp, which combines the prolonged biostability of plasmid DNA with the high transfection efficiency and gene silencing capabilities of siRNA.
RNA interference, a natural cell process in which specific mRNAs are targeted for degradation by complementary siRNAs, enables the specific silencing of a single gene at the cellular level.1, 2, 3 A variety of biomedical4 and clinical research1, 2, 5 has shown that RNA interference has great potential as a therapeutic approach. However, despite the tremendous potential of RNA interference for gene therapy and the large number of genes identified as candidates for targeting, success has been limited, largely because of complications associated with delivery. Synthetic siRNAs must be replenished constantly because they are destroyed along with their mRNA target during RNA interference-mediated gene silencing. Their destruction, combined with their susceptibility to environmental nucleases, limits their in vivo use.
Attenuated viruses achieve high cell transfection and intracellular delivery; however, concerns about the safety, immunogenicity and latent pathogenic effects of viral vectors have limited their therapeutic potential. In addition, there is concern that viral vectors will integrate into the genome and that they may recover the ability to cause disease. Several gene therapy clinical trials have been abandoned because of serious, in some cases fatal, consequences resulting from the use of viral vectors.6
DNA plasmid vector systems are a viable alternative for delivery of short hairpin RNAs (shRNAs) for gene-targeting therapy.3 DNAs are stable, shRNA expression can be inducible7 and targeted genes can be regulated for several months by maintaining selection for the plasmid.8 Limitations of conventional DNA vectors are primarily related to their size. Plasmids are typically >3000 bp and are difficult to minimize because of essential bacterial replication and selection sequences needed for vector propagation. Bacterial sequences contain immunotoxic CpG motifs9, which are approximately four times more prevalent in bacterial than mammalian DNA.10 Bacterial sequences can also induce transcriptional silencing of episomal transgenes11, 12, 13, 14, 15, 16 and may interfere with shRNA expression.
Reducing the size of DNA vectors appears to be a reasonable approach to improve cell transfection.17, 18 The use of minimized vectors appears to be especially effective for cell types that are refractory to transfection.17 For example, luciferase expression from a 2900 bp minicircle in aortic smooth muscle cells, which are difficult to transfect, was increased 77-fold relative to a 52 500 bp plasmid. The same 2900 bp minicircle yielded a 6-fold improvement in NIH 3T3 cells, which are far easier to transfect.
Results
A new DNA vector for gene therapy
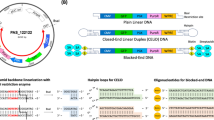
Minivector DNA can be as small as ∼250 bp and can be generated at high purity in milligram quantities.19 Obtaining large quantities of minicircles less than 500 bp was a technical challenge that was previously insurmountable, with micrograms quantities being the best achievable yields.20 Minivector DNAs contain only a promoter and the sequence-encoding shRNA and are nearly completely devoid of bacterial sequences (Figure 1). Along the convention of ‘p’ in front of plasmid names, we designate Minivector DNAs with ‘mv.’ Here, we compared biostability, cell transfection, gene silencing capacity and therapeutic potential of Minivector DNA, conventional DNA plasmid vectors and synthetic siRNA.

Minivector DNA construction. Minivector DNA for expression of shRNA against GFP and ALK were generated using the λ-integrase (Int) site-specific recombination system as described in Fogg et al.19 shRNA sequences were cloned into the parent plasmid (far left); the three parent plasmids (pMV) and Minivector (mv) DNAs used in this study are identified.
Biostability
Minivector and plasmid DNAs encoding shRNA or synthetic siRNA were incubated in 100% human serum at 37 °C. At each time point, as indicated in Figure 2a, residual DNA vectors and siRNA oligonucleotides were extracted and analyzed by gel electrophoresis. The Minivector DNA was stable in human serum for >48 h, whereas the parental plasmid DNA was more than 50% degraded by ∼4 h. The synthetic siRNA was degraded in less than 30 min (Figure 2b). Serum survival is an important first step toward in vivo use of Minivector DNA for gene therapy.

Nucleic acid stability in human serum. (a) Ethidium bromide-stained gels of Minivector DNA, plasmid and synthetic siRNA are shown. M, linear DNA markers in base pairs (bp). (b) Data from (a) are plotted. The lines shown are best fit to the data points. The experiment was repeated with similar results.
Transfection/gene silencing
We used two different cell lines stably expressing green fluorescent protein (GFP) to compare transfection and gene silencing mediated by plasmid, Minivector DNA or siRNA: adhesion 293FT cells, a transformed human embryonic kidney fibroblast cell line that is easily transfected, and suspension Jurkat cells, a human lymphoma/leukemia cell line that is difficult to transfect. shRNA against GFP was encoded on plasmid or Minivector DNA under control of the HI promoter. siRNA targeting GFP was synthesized. The three nucleic acids were separately transfected into cells using Lipofectamine (Invitrogen, Carlsbad, CA, USA). After 3 days, GFP was quantified by flow cytometry.
Figure 3a shows that the nucleic acids with control sequences had no effect on GFP expression. Plasmid, Minivector DNA and siRNA against GFP induced significant GFP gene silencing (34, 52 and 58%, respectively) in the adhesion 293FT cells (Figure 3b). In the Jurkat lymphoma/leukemia cells (Figure 3c), the plasmid had little effect on suppressing GFP expression (∼4.5%), as expected for a difficult to transfect cell line. In contrast, Minivector DNA (∼46%) was nearly as effective as synthetic siRNA (∼61%) in silencing the GFP gene in Jurkat cells. These data show that despite the very small size of Minivector DNA, RNA polymerase (presumably RNA polymerase III because it is the HI promoter) must find and transcribe shRNA from it. Unlike the synthetic siRNA, which will be degraded with gene silencing if it could survive delivery, Minivector DNA continues to deliver the transcribed shRNA.

Cell transfection/gene-silencing capacity. Changes in mean fluorescence intensity (%) of cellular GFP in cells treated as indicated were calculated relative to the untreated cells. (a) Control vectors transfected into 293 FT cells: pMV-H1 includes the H1 promoter but lacks the shRNA-expressing sequence; mvCCR5shRNA3 is a Minivector DNA expressing shRNA against the CCR5 gene and is a control for non-specific effects; control siRNA was provided by the manufacturer (Ambion). Plasmid, Minivector DNA or siRNA targeted against GFP transfected into (b) 293FT cells and (c) GFP (+) Jurkat cells. These experiments were repeated twice with the same results.
Anaplastic large cell lymphoma (ALCL) cells carry a chromosomal translocation resulting in abnormal expression of the anaplastic lymphoma kinase (ALK) gene.21 The auto-activation of ALK fusion protein22 is a key pathogenesis factor for ALCL development.23, 24, 25 siRNA-induced ALK gene silencing can inhibit the growth of ALCL cells.26, 27 We compared the ability of plasmids, Minivector DNA and synthetic siRNA in silencing ALK expression in cultured Karpas 299 cells (a human ALCL cell line from the German resource center for biological material (DSMZ)), and in parallel assessed their ability to inhibit ALCL cell growth. ALK was quantified using flow cytometry and an FITC-conjugated anti-ALK antibody. Minivector DNA silenced ALK fusion protein expression in Karpas 299 cells to ∼25%, the same extent as synthetic siRNA (Figure 4a), whereas, as expected, conventional plasmid vector caused, perhaps, a ∼1% decrease of ALK expression. An MTT cell proliferation assay revealed that transfection of Karpas 299 cells with Minivector DNA or synthetic siRNA resulted in an ∼40% inhibition of cell growth relative to control cells carrying the transfection vehicle (P<0.01, Figure 4b). Transfection of control nucleic acids or plasmid encoding shRNA against ALK had no detectable effect on cell growth.

(a) ALK gene silencing in and (a) growth arrest (b) of ALCL cells. ALK expression was determined using FITC-conjugated anti-ALK antibody. Gene silencing was quantified by the change in mean fluorescence intensity (%). (b) Relative cell growth (%) was calculated relative to the untreated cells as a background control. The experiments were performed three times and the results for (b) are presented as the mean±s.d. **P<0.01.
Discussion
Many human cell lines, for example, lymphoma cells, dendritic cells and T-cells, cannot be efficiently transfected with current plasmid vectors. The data presented here establish the utility of Minivector DNA as a tool to deliver shRNA to cells, including cells that are refractory to transfection with plasmids. In addition to being a useful tool for basic research, Minivector DNAs hold promise as a gene therapy tool in the clinic.
With a validated target, ALCL is a disease with high potential for RNA interference-based therapy. However, although siRNA works in cell culture, it is not stable in serum, requires constant replenishment and is prohibitively expensive, all but ruling out its therapeutic potential. Plasmid vectors do not efficiently transfect lymphoma cells and are therefore unsuitable. With high gene silencing capability and biostability, Minivector DNA overcomes these problems.
Minivector DNA survival in serum suggests that they may be stable following hematogenous delivery, allowing them to reach the target cells. Our work here is an important first step in establishing the therapeutic potential of Minivector DNA, particularly for a blood-borne disease such as lymphoma. To further increase the efficacy of Minivector DNA and to reduce potential off-target effects, ongoing and future studies will investigate ways to deliver Minivector DNA specifically to lymphoma cells.
The use of siRNA-based gene silencing has been an extremely useful tool for delineating the molecular processes causing many diseases, but the potential of clinical siRNA-based therapy is low. Minivector DNA represents a technological advance in delivery for gene therapy and holds great promise for use in a wide range of basic research and clinical settings.
Materials and methods
Minivector DNA generation
shRNA sequences were cloned as BglII–HindIII fragments into the parent plasmid, pMVCCR5shRNA3-BglII which was generated as follows. KasI and HindIII restriction sites were engineered into pMC339-BbvCI19 (QuikChange site-directed mutagenesis kit, Stratagene, La Jolla, CA, USA). The KasI/HindIII restriction fragment containing the H1 promoter and shRNA sequence from pSUPER-CCR5shRNA-38, 28 was inserted between the KasI and HindIII sites of pMC339-BbvCI. A BglII site was engineered in front of the shRNA expression sequence using the QuikChange kit to generate pMV-CCR5shRNA3-BglII. DNA inserts encoding shRNA against GFP29 and ALK30 were subcloned into pMV-CCR5shRNA3-BglII between the BglII and HindIII sites to generate pMV-H1-GFPshRNA and pMV-H1-ALKshRNA. Minivector DNA parent plasmids were transformed into E. coli strain LZ5431 and large-scale in vivo λ-integrase (Int)-mediated recombination and Minivector DNA isolation was performed as described previously with the following minor modification. Pure, monomeric supercoiled DNA was isolated by multiple rounds of Sephacryl S-500 gel-filtration (GE Healthcare Life Sciences, Piscataway, NJ, USA) instead of the preparative gel electrophoresis step. Fractions containing supercoiled, monomeric minicircle were pooled, concentrated using Amicon Ultra centrifugal filters (Millipore, Billerica, MA, USA), precipitated with ethanol and resuspended in 10 mM Tris-HCl, pH 8.0 and stored at −20 °C.
Nucleic acid stability in human serum
A total of 1 μg of plasmid, Minivector DNA or synthetic siRNA were incubated at 37 °C in 100 μl of 100% human serum (Atlanta Biological, Lawrenceville, GA, USA). At the time points shown (Figure 2), residual DNA vectors or siRNA were extracted with phenol:chloroform:isoamyl alcohol (25:24:1), extracted with chloroform, precipitated in 95% ethanol and analyzed on agarose gels (DNAs) or polyacrylamide gels (siRNA), followed by ethidium bromide staining. The bands of DNA or siRNA products were quantified using TotalLab software (FotoDyne, Hartland, WI, USA) and plotted using Kaleidagraph (Synergy Software, Reading, PA, USA).
Cell transfection/gene silencing
As a reporter system for GFP gene silencing, we established stable GFP-expressing cells from adhesion 293FT cells (a transformed human embryonic kidney cell line, Invitrogen) or suspension Jurkat cells (a human T-lymphoma/leukemia cell line from ATCC, Manassas, VA, USA). The GFP-expressing cells were transfected with 1 μg of Minivector DNA or plasmid (given the difference between the sizes, ∼10-fold more moles of Minivector are delivered relative to plasmid) or 20 pmol synthetic siRNAs using Lipofectamine methodology following the manufacturer's instructions (Invitrogen). Synthetic siRNAs against ALK were purchased with paired control siRNA (catalog #AM4626 from Ambion, Foster City, CA, USA). Silencing of cellular GFP in 293FT cells and Jurkat cells was quantified using flow cytometry at day 3 using a cell permeabilization kit from BD Biosciences (San Jose, CA, USA) according to the manufacturer's protocol. Data were analyzed with FlowJo software (BD Biosciences). The siRNA for silencing the ALK gene was synthesized by Ambion with the sense: 5′-CACUUAGUAGUGUACCGCCtt-3′ and antisense: 5′-GGCGGUACACUACUAAGUGtt-3′sequences previously shown to silence ALK.30 At 4 days after transfection, cellular ALK fusion protein levels were quantified by flow cytometry with FITC-conjugated anti-ALK antibody and calculated by mean fluorescence intensity.
Inhibition of lymphoma cell growth
Aliquots from the transfected Karpas 299 cells (100 μl per sample) were transferred to a 96-well plate, mixed with 10 μl of assay buffer from the MTT kit from Chemicon International (Temecula, CA, USA), incubated at 37 °C for 4 h and lysed per the manufacturer's instructions. The MTT cell proliferation assay was analyzed using a BioRad (Hercules, CA, USA) microplate reader by optical density at OD540.
References
Kim B, Tang Q, Biswas PS, Xu J, Schliffelers RM, Xie FY et al. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am J Pathol 2004; 165: 2177–2185.
Haussecker D, Cao D, Huang Y, Parameswaran P, Fire AZ, Kay MA . Capped small RNAs and MOV10 in human hepatitis delta virus replication. Nat Struct Mol Biol 2008; 15: 714–721.
Shi Y . Mammalian RNAi for the masses. Trends Genet 2003; 19: 9–12.
Taroncher-Oldenburg G, Marshall A . Trends in biotech literature 2006. Nat Biotechnol 2007; 25: 961.
Kurreck J . RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed Engl 2009; 48: 1378–1398.
Anonymous. Gene therapy deserves a fresh chance. Nature 2009; 461: 1173.
Ma HT, On KF, Tsang YH, Poon RY . An inducible system for expression and validation of the specificity of short hairpin RNA in mammalian cells. Nucleic Acids Res 2007; 35: e22.
Brummelkamp TR, Bernards R, Agami R . A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550–553.
Bigger BW, Tolmachov O, Collombet JM, Fragkos M, Palaszewski I, Coutelle . An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J Biol Chem 2001; 276: 23018–23027.
Swartz MN, Trautner TA, Kornberg A . Enzymatic synthesis of deoxyribonucleic acid. XI. Further studies on nearest neighbor base sequences in deoxyribonucleic acids. J Biol Chem 1962; 237: 1961–1967.
Chen ZY, He CY, Meuse L, Kay MA . Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Therapy 2004; 11: 856–864.
Darquet AM, Cameron B, Wils P, Scherman D, Crouzet J . A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Therapy 1997; 4: 1341–1349.
Yew NS, Zhao H, Przbylska M, Wu IH, Tousignant JD, Scheule RK et al. CpG-depleted plasmid DNA vectors with enhanced safety and long-term gene expression in vivo. Mol Ther 2002; 5: 731–738.
Reyes-Sandoval A, Ertl HC . CpG methylation of a plasmid vector results in extended transgene product expression by circumventing induction of immune responses. Mol Ther 2004; 9: 249–261.
Chen ZY, He CY, Ehrhardt A, Kay MA . Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther 2003; 8: 495–500.
Mamanova L, Andrews RM, James KD, Sheridan EM, Ellis PD, Langford CF et al. FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nat Methods 2010; 7: 130–132.
Kreiss P, Cameron B, Rangara R, Mailhe P, Aguerre-Charriol O, Airiau M et al. Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency. Nucleic Acids Res 1999; 27: 3792–3798.
Yin W, Xiang P, Li Q . Investigations of the effect of DNA size in transient transfection assay using dual luciferase system. Anal Biochem 2005; 346: 289–294.
Fogg JM, Kolmakova N, Rees I, Magonov S, Hansma H, Perona JJ et al. Exploring writhe in supercoiled minicircle DNA. J Phys Condens Matter 2006; 18: S145–S159.
Bednar J, Furrer P, Stasiak A, Dubochet J, Egelman EH, Bates AD . The twist, writhe and overall shape of supercoiled DNA change during counterion-induced transition from a loosely to a tightly interwound superhelix. Possible implications for DNA structure in vivo. J Mol Biol 1994; 235: 825–847.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994; 263: 1281–1284.
Ladanyi M . The NPM/ALK gene fusion in the pathogenesis of anaplastic large cell lymphoma. Cancer Surv 1997; 30: 59–75.
Piva R, Chiarle R, Manazza AD, Taulli R, Simmons W, Ambrogio C et al. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood 2006; 107: 689–697.
Amin HM, Lai R . Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007; 110: 2259–2267.
Ritter U, Damm-Welk C, Fuchs U, Bohle RM, Borkhardt A, Woessmann W . Design and evaluation of chemically synthesized siRNA targeting the NPM-ALK fusion site in anaplastic large cell lymphoma (ALCL). Oligonucleotides 2003; 13: 365–373.
Hsu FY, Zhao Y, Anderson WF, Johnston PB . Downregulation of NPM-ALK by siRNA causes anaplastic large cell lymphoma cell growth inhibition and augments the anti cancer effects of chemotherapy in vitro. Cancer Invest 2007; 25: 240–248.
Marzec M, Kasprzcka M, Ptasznik A, Wlodarski P, Zhang Q, Odum N et al. Inhibition of ALK enzymatic activity in T-cell lymphoma cells induces apoptosis and suppresses proliferation and STAT3 phosphorylation independently of Jak3. Lab Invest 2005; 85: 1544–1554.
Poluri A, Sutton RE . Titers of HIV-based vectors encoding shRNAs are reduced by a dicer-dependent mechanism. Mol Ther 2008; 16: 378–386.
Tiscornia G, Singer O, Ikawa M, Verma IM . A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA 2003; 100: 1844–1848.
Ito M, Zhao N, Zeng Z, Chang CC, Zu Y . Synergistic inhibition of anaplastic large cell lymphoma cells by combining cellular ALK gene silencing and a low dose of the kinase inhibitor U0126. Cancer Gene Ther 2010; 17: 633–644.
Zechiedrich EL, Khodursky AB, Cozzarelli NR . Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in Escherichia coli. Genes Dev 1997; 11: 2580–2592.
Acknowledgements
We thank Dr Ana Poluri and Dr Richard Sutton for pSUPER-CCR5shRNA-3 and for helpful suggestions and advice, and Dr Mary-Jane Lombardo for critically reading the manuscript. JMF was a fellow of the Program in Mathematics and Molecular Biology of Florida State University supported by the Burroughs Welcome Fund. This work was supported by a National Institutes of Health (NIH) grant (K22CA113493) and a TMHRI CTS Award to YZ and NIH grant RO1A1054830 to LZ.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Dr Jonathan Fogg and Dr Lynn Zechiedrich are co-inventors on an issued patent and another patent application, which cover the technology that was used in the work reported in this paper.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhao, N., Fogg, J., Zechiedrich, L. et al. Transfection of shRNA-encoding Minivector DNA of a few hundred base pairs to regulate gene expression in lymphoma cells. Gene Ther 18, 220–224 (2011). https://doi.org/10.1038/gt.2010.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2010.123
Keywords
This article is cited by
-
Base-pair resolution analysis of the effect of supercoiling on DNA flexibility and major groove recognition by triplex-forming oligonucleotides
Nature Communications (2021)
-
Supercoiling and looping promote DNA base accessibility and coordination among distant sites
Nature Communications (2021)
-
Gold Nanoparticles for Nucleic Acid Delivery
Molecular Therapy (2014)
-
Micro-minicircle Gene Therapy: Implications of Size on Fermentation, Complexation, Shearing Resistance, and Expression
Molecular Therapy - Nucleic Acids (2014)
-
Supercoiled Minivector DNA resists shear forces associated with gene therapy delivery
Gene Therapy (2012)
