
Abstract
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) is a disorder of development of the vasculature characterized by telangiectases and arteriovenous malformations in specific locations. It is one of most common monogenic disorders, but affected individuals are frequently not diagnosed. The most common features of the disorder, nosebleeds, and telangiectases on the lips, hands, and oral mucosa are often quite subtle. Optimal management requires an understanding of the specific presentations of these vascular malformations, especially their locations and timing during life. Telangiectases in the nasal and gastrointestinal mucosa and brain arteriovenous malformations generally present with hemorrhage. However, complications of arteriovenous malformations in the lungs and liver are generally the consequence of blood shunting through these abnormal blood vessels, which lack a capillary bed and thus result in a direct artery-to-vein connection. Mutations in at least five genes are thought to result in hereditary hemorrhagic telangiectasia, but mutations in two genes (ENG and ACVRL1/ALK1) cause approximately 85% of cases. The frequency of arteriovenous malformations in particular organs and the occurrence of certain rare symptoms are dependent on the gene involved. Molecular genetic testing is used to establish the genetic subtype of hereditary hemorrhagic telangiectasia in a clinically affected individual and family, and for early diagnosis to allow for appropriate screening and preventive treatment.
Similar content being viewed by others


INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT) is characterized by the presence of multiple arteriovenous malformations (AVMs) that lack intervening capillaries and result in direct connections between arteries and veins. Small AVMs are called telangiectases. Telangiectases are most evident on the lips, tongue, face, and fingers, and the nasal, buccal, and gastrointestinal (GI) mucosa. They appear as pink to red, pinpoint to pinhead-size lesions, or occasionally as larger, even raised purple lesions (Fig. 1). Telangiectases are distinguished from petechiae and angiomata by their ability to blanch with pressure and then immediately refill. Because of their thin walls, narrow tortuous paths, and proximity to the surface of the skin or to a mucous membrane, telangiectases can rupture and bleed, sometimes with minimal or no trauma. As the contractile elements in the vessel wall are lacking, given the direct arterial connection, bleeding from telangiectases can be brisk and difficult to stop.

Telangiectases of the (A) lips and tongue; (B) fingers—note typical, subtle lesions on the distal 2nd and 3rd digits; and (C) nasal mucosa.
The term AVM usually refers to the “large” telangiectases, greater than a few millimeters in diameter and sometimes up to several centimeters in diameter. AVMs occur most commonly in the liver, lung, or brain (Fig. 2). In the brain, they are typically present at birth, whereas AVMs may develop or grow over time in the lung and liver. In contrast to the complications of smaller telangiectases, the complications of AVMs often result from the shunting of blood, leading to increased cardiac output and, in the lung, desaturation of arterial blood. Lung AVMs also provide a ready right-to-left shunt for venous emboli (clots or bacteria) to reach the arterial circulation. Occasionally, a pulmonary AVM (PAVM) can rupture, leading to hemoptysis. Although HHT is a usually progressive disorder, infants are occasionally severely affected with complications of pulmonary or cerebral AVMs. However, in most affected individuals, the features are age dependent with the diagnosis not suspected until adolescence or later.

Arteriovenous malformations of the (A) lungs by CT and (B) brain by MRI.
The original descriptions of familial epistaxis were made by Sutton in 18641 and Babington in 1865.2 However, Osler in the United States, Parkes Weber in the United Kingdom, and Rendu in France are typically given credit, hence the eponymous Osler-Weber-Rendu syndrome or Rendu-Osler-Weber. The designation “hereditary hemorrhagic telangiectasia” was suggested in 1912 by one of Osler's students.
HHT is a heterogenous autosomal dominant condition in which de novo mutations are rare. Most individuals diagnosed with HHT are determined to have an affected parent on targeted physical examination and medical history. The overall prevalence of HHT in North America is estimated at 1:10,0003; however, it is likely that this is an underestimate.4 When a proband is diagnosed, it is not unusual to identify multiple relatives in the family who have not been labeled with HHT but who have struggled with epistaxis, embolic stroke, or GI bleeding. HHT occurs with wide ethnic and geographic distribution.
CLINICAL DESCRIPTION
HHT displays age-related penetrance with increased manifestations usually developing over a lifetime. The average age for development and/or symptomatic presentation of AVMs is very organ specific. In general, the disorder is more often progressive with respect to the smaller lesions of the dermis and mucosa. However, observable manifestations can be subtle even well into adulthood. For example:
-
Approximately 50% of diagnosed individuals report having nosebleeds by the age of 10 years and 80–90% by the age of 21 years. As many as 95% eventually develop recurrent epistaxis.5,6
-
The percentage of individuals with telangiectases of the hands, face, and oral cavity is similar to the percentage with epistaxis, but the age of onset of visible telangiectases is generally 5–30 years later than for epistaxis.7,8
-
Cerebral AVMs are overwhelmingly congenital, and intracranial hemorrhage secondary to cerebral AVM has been reported as the presenting symptom of HHT in infants and children with HHT.9
Epistaxis
Nasal bleeding is the feature that most commonly brings patients with HHT to medical attention. Average age of onset is approximately 12 years but ranges from infancy to adulthood. Although nosebleeds are often spontaneous, they also occur more often in patients with HHT secondary to minimal trauma to the nose. Nosebleed frequency in affected adults ranges from several per year to several per day. Nosebleed severity ranges from a couple drops of bright red blood that collects within the nose, to gushing. Many affected individuals have only mild, infrequent nosebleeds that never require medical attention, and thus, the nose bleeding can be difficult to elicit when taking a medical or family history. Many of those with HHT whose nosebleeds are on the mild end of the spectrum initially answer “no” when asked about recurrent nosebleeds, assuming that recurring mild nosebleeds are “normal.” In a minority of patients, epistaxis leads to chronic anemia and transfusion dependence.
Telangiectasia
Although similar proportions (95%) of affected individuals eventually develop telangiectases of the face, oral cavity, or hands, the average age of recognition is generally later than epistaxis but may be during childhood. Thirty percent of affected individuals report telangiectases first appearing before the age of 20 years and two thirds before the age of 40 years.6 However, on careful examination, most affected individuals have several telangiectases in characteristic locations within the first decade of life. Even well into adulthood, the number of observable telangiectases in affected individuals often number only 5–15 and can be quite subtle. The telangiectases of HHT are rarely diffuse and often not particularly “striking” except in some older patients. Adults who do not have HHT can accumulate cutaneous telangiectases, especially in sun-exposed areas, as they age. The telangiectases of chronic liver disease are of the “spider” variety, with a central core and small vessels radiating outward. In distinction, most telangiectases in HHT are punctuate or macular. Telangiectases can be found anywhere in the GI system, but most commonly, the stomach and the proximal small intestine (duodenum and jejunum) are involved.
GI bleeding
Approximately one quarter of all adults with HHT eventually have GI bleeding, and the responsible telangiectases are overwhelmingly in the upper GI tract.10–13 Bleeding from GI telangiectases most commonly begins after the age of 50 years, is usually slow but persistent, and often becomes increasingly severe with age. No particular foods or activities have been identified as contributors to GI bleeding in individuals with HHT.
Pulmonary
The prevalence of PAVMs in patients with HHT is dependent on the method used for detection. In one recent study, 37% of patients with HHT had at least one AVM demonstrable by high-resolution computed tomography (CT). However, 59% had evidence of a pulmonary shunt by transthoracic contrast echocardiography (TTCE) with agitated saline contrast.14 In the past decade, TTCE (commonly called echo bubble) has been shown to be a sensitive screen for PAVMs15 and has become the standard initial screen in patients with HHT.16 Only those with a contrast echocardiogram suggestive of pulmonary shunting are exposed to the radiation of chest CT. There is current debate about whether a minor degree of shunting detected by echo bubble deserves CT follow-up, as most radiographic studies in such patients fail to detect an AVM. Indeed, studies now document small-to-moderate shunts by TTCE in otherwise healthy individuals17 and that shunts increase with exercise.18 However, antibiotic prophylaxis for dental work, and other procedures with a significant risk for bacteremia, is recommended for all patients with HHT with evidence of a pulmonary shunt by contrast echocardiogram—whether the AVM(s) can be seen by chest CT.
In one series, serious neurologic events including transient ischemic attack, stroke, and brain abscess occurred in 30–40% of individuals with PAVMs with feeding arteries 3.0 mm or greater in diameter.19 Another study found that the risk of ischemic stroke secondary to PAVM was significant and was not associated with any marker of AVM severity, such as feeder artery size.20 Neurologic complications may occur in individuals with near-normal arterial oxygen tension and no complaint of dyspnea. Thus, screening for asymptomatic PAVMs is warranted. PAVMs frequently enlarge with time.19
Pulmonary hypertension is another vascular manifestation of HHT. Although much less common than PAVMs, it can result from either systemic arteriovenous shunting in the liver increasing cardiac output or be clinically and histologically indistinguishable from idiopathic pulmonary arterial hypertension.21
Central nervous system
Cerebral AVMs occur in approximately 10% of affected individuals.22,23 The risk of cerebral hemorrhage from these high-flow lesions is generally considered to warrant treatment of even asymptomatic individuals of young age. The size, morphology, and location of a particular lesion factor into the decision making about treatment.
The most common central nervous system insults in patients with HHT, brain abscess, and ischemic stroke are actually secondary to the right-to-left shunting associated with PAVMs.
Spinal AVMs are significantly less common than brain AVMs and usually present with paralysis and/or complaint of back pain. Most are diagnosed and treated in the first decade of life.
Hepatic
The frequency of hepatic vascular abnormalities was 74% in one study that systematically imaged the liver of affected individuals using CT24 and 41% in another study using ultrasound examination.25 However, only a small minority (8% in the study using CT) were symptomatic. Hepatic focal nodular hyperplasia occurs in HHT at a prevalence greater than the general population,26 and the radiologic imaging may raise concerns of a hepatic tumor. Because needle biopsy of the liver is contraindicated in HHT because of the risk of hemorrhage, recognizing that focal nodular hyperplasia is a much more likely diagnosis than cancer will help guide management.
CLINICAL DIAGNOSIS
The clinical diagnosis of HHT is based on the presence of multiple AVMs and/or telangiectasia in characteristic locations.3,4 Consensus diagnostic criteria—often referred to as the Curacao criteria—were published in 2000.27
Findings
-
Epistaxis (nosebleeds): spontaneous and recurrent.
-
Mucocutaneous telangiectases: multiple, at characteristic sites (lips, oral cavity, fingers, and nose). Lesions are blanchable and usually punctuate, pink-red in color, and pinpoint to pinhead in size. Occasionally 2–5 mm macules, purple or “spidery.”
-
Visceral AVM.
-
Pulmonary
-
Cerebral
-
Hepatic
-
GI
-
Spinal
-
Family history: a first-degree relative in whom HHT has been diagnosed using these criteria.
The clinical diagnosis of HHT is considered:
-
Definite when three or more findings are present;
-
Possible or suspected when two findings are present; and
-
Unlikely when fewer than two findings are present.
Note: These diagnostic criteria were established for adults and can be misleading when applied to children. Symptoms and signs of HHT generally develop during childhood and adolescence, such that the absence of epistaxis, telangiectases, or symptoms of solid organ AVM is common in affected children.9,28 Children with an affected parent should undergo molecular genetic testing to confirm or refute the diagnosis.29
The appearance of telangiectases at characteristic sites (lips, oral cavity, hands, and nasal septum) is key. If patient has an isolated visceral vascular lesion, but not these, HHT is very unlikely.
Most individuals with a PAVM(s) have HHT.30 In contrast, cerebral AVMs occur most frequently as an isolated finding but may be a manifestation of HHT or another dominantly inherited vascular dysplasia such as capillary malformation-AVM caused by mutations in RASA1.31 A family history of recurrent nosebleed and presence of telangiectases specifically on the lips, face, and hands help distinguish HHT from other vascular dysplasias or from an isolated AVM.
A rare syndrome has been identified that combines HHT and juvenile polyposis (HHT/JP syndrome) and results from mutations in SMAD4.32 It is currently thought to account for approximately 2% of HHT.33,34 Thus, family and medical history should include questions about the occurrence of GI polyps and cancer when HHT is known or suspected.
MEDICAL MANAGEMENT OVERVIEW
Although hemorrhage is usually the presenting symptom of mucosal telangiectases and cerebral AVMs, most visceral AVMs present as a consequence of blood shunting through the abnormal vessel and bypassing the capillary bed. Shunting of air, thrombi, and bacteria through PAVMs, thus bypassing the filtering capabilities of the lungs, may cause transient ischemic attacks, embolic stroke, and cerebral and other abscesses. Migraine headache, polycythemia, and hypoxemia with cyanosis and clubbing of the nails are other complications of PAVMs.21,23,35 Hepatic AVMs can present as high-output heart failure, portal hypertension, or biliary disease.36
Optimal medical management for HHT requires distinguishing between organ locations where telangiectases and AVMs are best managed symptomatically/expectantly, versus those in which lesions should be detected and treated before the onset of symptoms. International Clinical Management Guidelines for HHT were published as a result of a consensus conference.29 In general, telangiectases of the skin, oral and GI mucosa, and liver are treated when symptoms dictate, but AVMs of the lungs and brain are treated in patients without symptoms given their often sudden and catastrophic presentation. This distinction determines routinely recommended surveillance for patients with HHT. Most of the recommendations were based on expert opinion and very few on randomized clinical trials. Additionally, variations in access to imaging modalities resulted in disagreement about best practices in HHT Centers of Excellence in different parts of the world. The HHT Foundation International (www.hht.org) lists HHT Centers in the United States and elsewhere, as well as information regarding current management for both patients and clinicians.
SURVELLIANCE/SCREENING
Initial evaluation
To initially establish the extent of disease in an individual diagnosed with HHT, the following evaluations are recommended:
-
Medical history, with particular attention to epistaxis and other bleeding, anemia or polycythemia, diseases of the heart, lung and liver, and neurologic symptoms.
-
Physical examination, including inspection for telangiectases, particularly on fingers, lips, tongue, oropharynx, cheeks, or conjunctiva, as well as listening for bruits over the liver.
-
Complete blood count and serum ferritin, with particular attention to anemia, polycythemia, and the need for iron supplementation. If anemia is present, it is important to determine whether the anemia seems to be disproportionate to the amount of epistaxis.
-
In family index case-molecular genetic testing to determine family HHT type. A causative mutation identified in one of the two common HHT genes (ENG or ACVRL1) provides reassurance that the added risk of juvenile polyposis conferred by a mutation in SMAD4 is not a concern. If no mutation is identified in ENG or ACVRL1, testing of SMAD4 should be considered.29,33
-
Measurement of oxygen saturation by pulse oximetry.
-
Contrast echocardiography for detection of pulmonary arteriovenous shunting37,38 and measurement of the pulmonary artery systolic pressure as a screen for pulmonary artery hypertension.39 When pulmonary shunting is suggested, high-resolution chest CT to define size of lesions(s) is the next step.15,40 Some, but not all, HHT Centers prefer to use contrast. If negative, the conservative approach is to perform a screening chest CT every 5 years for life; however, the risk of developing a new PAVM after mid life is probably exceedingly small. The exact recommended follow-up interval and modality should be determined with consideration to age and findings at last evaluation.29
-
Head magnetic resonance imaging (MRI, with and without gadolinium) to detect cerebral AVMs as early as possible, preferably in the first 6 months of life. If no cerebral AVMs are detected by this scan in adulthood, no further screening for cerebral AVMs is suggested.29
-
Consideration of abdominal ultrasound or CT examination to look for evidence of hepatic AVM, if the individual has symptoms of hepatic involvement such as high-output failure or a bruit over the right upper quadrant of the abdomen.
Note: (1) Screening for hepatic AVMs in asymptomatic individuals is not common practice because hepatic AVMs are rarely symptomatic and, when they do become symptomatic, it is not sudden and catastrophic, as is seen with PAVMs and cerebral AVMs. (2) Also, current treatment options for hepatic AVM are less satisfactory.
Follow-up evaluation
The following protocol is recommended for follow-up of all individuals for whom the diagnosis of HHT is definite, and for all individuals at risk for HHT based on family history, in whom HHT has not been ruled out by molecular diagnosis:
-
Annual evaluation by a healthcare provider familiar with HHT, including interval history for epistaxis or other bleeding, shortness of breath or decreased exercise tolerance, and headache or other neurologic symptoms.
-
Periodic hematocrit/hemoglobin with appropriate treatment for anemia.
-
Reevaluation for PAVM at approximately 5-year intervals. Contrast echocardiogram is used if the previous contrast echocardiogram did not reveal evidence of a right to left shunt; chest CT is used if the previous contrast echocardiogram revealed evidence of a right to left shunt.
-
Screening for GI polyps and malignant change in persons with SMAD4 mutations.
Childhood
Because serious complications of pulmonary and cerebral AVMs can occur at any age,9,28,30,31 and safe and effective treatment options exist,42 evaluation for pulmonary and cerebral AVMs is recommended in children with possible or definite HHT. Intracranial hemorrhage has been reported in the first several months of life in affected children with previously asymptomatic cerebral AVMs. However, most serious complications of PAVMs have occurred in hypoxemic children. Also, reperfusion of PAVMs treated in first decade of life is more common than when treated in adulthood.42 These observations guide the recommended timing and modality of surveillance in children.
-
Head MRI with and without gadolinium is recommended as early as the first few months of life. Although cerebral AVMs are thought to be overwhelmingly congenital, and repeat screening for cerebral AVMs in adults is generally not recommended, there is less consensus about whether an additional scan in adulthood is indicated when the initial brain screening was performed in childhood. Several as of yet unpublished cases document a cerebral AVM not apparent by contrast MRI in the first decade of life evident in early adulthood. In these few cases, it is possible that the sensitivity of the imaging 20 years after the initial study was improved enough to detect small lesions that, in reality, were present in childhood. However, this has made some recommend one repeat head MRI in adulthood if the initial one was performed in the first decade of life.
-
Until age 10–12 years, pulse oximetry in the supine and sitting positions every 1–2 years during childhood is recommended as a minimum to screen for PAVMs. It may be of concern if the sitting value is even a few percentage points below that of the supine value. (As most PAVMs are in the lower lobes, many individuals with PAVMs have higher oxygen saturation when lying than when sitting because of the effect of gravity.) Oxygen saturations below 97% should be followed up with contrast echocardiography. At approximately age 10–12 years, all children with HHT should have contrast echocardiography, with a follow-up CT if positive.
Pregnancy
The majority of pregnancies in patients with HHT proceed uneventfully. The most common HHT-related complications in patients with HHT are hemorrhage and stroke during the third trimester of pregnancy related to untreated PAVMs.43 Intracranial hemorrhage20,44 and one case of high-output cardiac failure related to hepatic vascular malformations have also been reported.45 Screening and treatment as indicated for pulmonary and cerebral AVMs are ideally performed before pregnancy in all women with HHT. Most HHT experts recommend that pregnant women who have not had a recent evaluation for PAVMs be evaluated during pregnancy. Contrast echocardiogram to rule out evidence of a pulmonary shunt can be performed in the first trimester. If positive, chest CT, with abdominal shielding, should be delayed until the second trimester. Women discovered to have a significant size PAVMs can then be treated by transcatheter embolization during the second trimester.
Prevention of secondary complications
If contrast echocardiography is positive for pulmonary shunting, even if no PAVM is demonstrated by chest CT, a lifetime recommendation for prophylactic antibiotics in accordance with the American Heart Association protocol with dental cleaning and other procedures with risk for bacteremia is advised. This is because of the high risk of abscess, particularly brain abscess, associated with right to left shunting in patients with HHT.46–48 For the same reason, an air filter or extreme caution not to introduce air bubbles is recommended with intravenous lines.
Agents/circumstances to avoid
Anticoagulants such as warfarin and nonsteroidal antiinflammatory agents such as aspirin and ibuprofen that interfere with platelet function should be avoided unless required for treatment of other medical conditions. However, a patient's bleeding history should be considered when weighing the risks and benefits. In a patient with HHT with infrequent, mild nosebleeds and no history of GI bleeding, the concern regarding these exposures may be negligible.
Scuba diving should be avoided unless contrast echocardiography performed within the last 5 years was negative for evidence of a right-to-left shunt.
TREATMENT
The timing, mode, and indications for treatment of the telangiectases and AVMs of HHT are extremely organ dependent.
Nose
It is appropriate to consider intervention for nosebleeds in the case of anemia attributable to the nosebleeds or if an individual feels that the frequency or duration interferes significantly with normal activities. Worsening of epistaxis is associated with high-output cardiac failure, and aggressive management of the latter is essential.49 The following are reported to be helpful in the management of epistaxis:
-
Humidification and the daily application of nasal lubricants may be helpful.
-
Hemostatic products (gauze, sponge, or powder products) available over the counter help patients self-manage significant nosebleeds.
-
Laser ablation is usually recommended as the intervention for control of mild to moderate nosebleeds.50
-
Severe epistaxis, which has proven unresponsive to the above methods, is treated by septal dermoplasty,51 Young's nasal closure,52 or use of a nasal obturator.53 Surgical treatment for severe epistaxis in persons with HHT should be performed by surgeons who treat HHT regularly.
-
Most otolaryngologists experienced with treating individuals with HHT advise against electric and chemical cautery and transcatheter embolotherapy for treatment of recurrent nosebleeds in most situations.
-
A recent meta-analysis of studies of hormonal and antihormonal treatment concluded that estrogen-progesterone at doses used for oral contraception may reduce or eliminate bleeding in symptomatic HHT and is a reasonable initial option to consider for fertile women.54 Antifibrinolytic drugs such as tranexamic acid (Cyklokapron®) have been used with some success in select patients, but the associated risks are not well established.55,56 Other agents for which case reports document improvement include bevacizumab (Avastin®) and thalidomide. Controlled trials with these agents and standard therapy are needed.
GI tract
Treatment is unnecessary unless aggressive iron therapy has been ineffective in maintaining hemoglobin concentration in the normal range. Studies of fecal occult blood are nonspecific as blood swallowed from epistaxis gives a positive result.
-
Push enteroscopy, capsule endoscopy, mesenteric and celiac angiography, and radionuclide studies may be used to localize the source of bleeding and its type.
-
Endoscopic application of a heater probe, bicap, or laser is the mainstay of local treatment.
-
In case reports, hormonal treatment with estrogen-progesterone57 and bevacizumab58 have decreased transfusion needs.
-
Small bowel bleeding sites and larger vascular malformations can be removed surgically after they are identified by nuclear medicine studies.
Anemia
Even with optimal, available treatment, epistaxis and/or GI bleeding can result in mild to severe iron deficiency anemia, sometimes requiring iron replacement therapy or rarely, blood transfusion. Aggressive iron replacement therapy, to include parenteral delivery if needed to maintain iron stores, is considered preferable to relying on blood transfusions to manage anemia resulting from HHT-related hemorrhage. In general, a target ferritin level of at least 50, and ideally 100, is recommended for patients with HHT with a chronic anemia. Epistaxis is the most common cause of anemia in patients with HHT and is often overlooked as the primary cause as investigations for evidence of GI bleeding are undertaken.
Pulmonary AVMs
Any PAVM with a feeding artery >1-3 mm detected by chest CT should be considered for treatment by transcatheter embolization.59,60 Treatment of PAVMs is indicated for dyspnea, exercise intolerance, and hypoxemia but is most important for prevention of lung hemorrhage and the neurologic complications of brain abscess and stroke, even in those who are asymptomatic in terms of pulmonary function and oxygen saturation.61 Migraine headaches are associated with PAVMs in patients with HHT and often resolve or are significantly reduced after embolization.62 Long-term follow-up by chest CT is indicated after transcatheter occlusion of PAVMs because of reported recanalization and development or growth of untreated PAVMs.21 Usually a follow-up CT is done 6–12 months postocclusion, and if no reperfusion of treated AVM(s) or new PAVMs is noted, follow-up CT is generally recommended at 5-year intervals thereafter.
Cerebral AVMs
Cerebral AVMs >1.0 cm in diameter are usually treated using neurovascular surgery, embolotherapy, and/or stereotactic radiosurgery.63
Hepatic involvement
Treatment of cardiac failure or liver failure secondary to hepatic vascular malformations is currently problematic.64 Moreover, the apparent severity of vascular abnormalities found on CT scanning does not correlate with symptoms.65 Embolization of hepatic AVMs, which is successful for treatment of PAVMs, has led to lethal hepatic infarctions. Most patients with symptomatic hepatic involvement can be satisfactorily managed with intensive medical therapy66 aimed at managing high-output cardiac failure and hepatocyte dysfunction. Liver transplantation has been the standard treatment for those (usually older) individuals whose symptoms do not respond to medical management.66,67 A dramatic reduction of liver vascularity and normalization of cardiac output after treatment with bevacizumab have been reported in a patient with HHT who had been listed for liver transplant.68
Liver biopsy should be avoided in individuals with HHT.66
Pharmacologic therapies
As available surgical treatments for certain manifestations of HHT can be temporizing or ineffective (e.g., ablation of GI telangiectases), involve significant morbidity (liver transplant and nasal septal dermoplasty), or are temporary (nasal laser coagulation), safe and effective drug therapies have long been anticipated by patients with HHT. Oral or intranasal topical estrogens can be effective, although long-term therapy is limited to women. Raloxifene may increase expression of the ENG and ACVRL1 genes.69 Recent small series and cases reports have suggested therapeutic benefit from antiangiogenesis drugs such as bevacizumab58,68,70,71 and thalidomide,72 and the antifibrinolytic drug tranexamic acid,55 particularly for the treatment of severe GI bleeding, epistaxis, and symptomatic hepatic vascular malformations. Randomized trials of many such agents are in process.
PATHOGENESIS
Three HHT genes have been identified to date, and several others are suspected:
-
Endoglin (ENG) encodes a homodimeric transmembrane protein, which is a major glycoprotein of the vascular endothelium. This protein is a component of the transforming growth factor beta (TGFβ)-bone morphogenic protein (BMP) receptor complex, and it binds TGFB1 and TGFB3 with high affinity. It is expressed predominantly on endothelial cells, and on syncytiotrophoblasts, activated monocytes, and tissue macrophages.
-
Activin A receptor type II-like 1 (ACVRL1): the normal gene product is a cell-surface receptor for TGFβ superfamily ligands. It is expressed predominantly not only on endothelial cells71 but also on lung and placental cells.
-
SMAD4 encodes a 552-amino acid protein that functions as an intracellular signaling molecule in the TGFβ/BMP pathway. Mutations cause HHT/juvenile polyposis combined syndrome.
-
Additional as-yet-unknown HHT genes are suggested by linkage analysis in affected kindreds. Cole et al.74 reported a 6 Mb interval on the long arm of chromosome 5 (5q31.3–5q32) and Bayrak-Toydemir et al.75 reported a 7 Mb region on the short arm of chromosome 7 (7p14). In addition, there are some families that are seemingly unlinked to any of these five loci (unpublished data). One patient with HHT and pulmonary hypertension, with no mutation in ENG, ACVRL1 or SMAD4, was found to have a nonsense mutation in BMPR2.76
HHT is caused by a disturbance in the TGFβ signaling pathway. TGFβ signaling is important in the regulation of many cellular processes such as proliferation, differentiation, adhesion, and migration. The superfamily signaling pathway includes TGFβ ligand proteins, receptor proteins, activins, BMPs, and cytoplasmic signaling molecules such as Smads. ACVRL1 is a TGFβ type 1 receptor. ENG associates with different signaling receptors and modifies TGFβ1 signaling. ACVRL1 and ENG work together on cellular responses such as proliferation and adhesion. TGFβ signaling occurs through phosphorylation of TGFβ receptor 1 by TGFβ receptor II, which activates downstream signaling by phosphorylation of Smads on the membrane of the cells. A specific smad protein (SMAD4) transfers this information to the nucleus and regulates transcription of the target gene(s).
Analysis of mice carrying mutations in either ENG or ACVRL1 has provided insight into the molecular mechanisms through which they act. Mouse embryos lacking either ENG or ACVRL1 develop AVMs and other HHT features.77,78 Mouse models showed that ACVRL1/ENG signaling is required for the development and maintenance of arteriovenous identity.79 In addition, ACVRL1 and ENG also seem to coordinate smooth muscle cell recruitment to developing vessels. Mice lacking ACVRL177,80 or ENG78,81,82 have an overall reduction of vascular smooth muscle cells (vSMCs), in addition to a disturbance of the recruitment of vSMCs to the vascular plexus in the yolk sac and embryo. Thus, ENG and ACVRL1 are necessary for recruitment of vSMC and the differentiation of vSMC, during the development of vascular system. Mutations in HHT genes lead to perturbation of vascular remodeling and the maintenance of vessel wall integrity.83
Current data suggest that most disease-causing mutations in ACVRL1 or ENG result in haploinsufficiency.84–86 Thus, HHT is assumed to result from lack of sufficient protein for normal function.73 Mutations resulting in structural alterations by misfolding and intracellular degradation of these proteins lead to lack of surface expression of the mutant proteins.
Mutations in HHT
There are no common disease-causing mutations or mutation hot spots in any of the HHT genes.
ENG mutations that cause HHT are dispersed almost equally throughout the gene with the exception of exons 1, 10, 14a, and 14c, which seem to have fewer mutations. Mutations of all types have been reported.73,87 Mutations causing sequence changes represent 85–90% of ENG mutations, and large deletions and/or duplications involving single or several exons represent 6–10% of mutations.88
Similarly, mutations have been identified in all exons of ACVRL1; however, mutations in exons 8, 7, and 3 account for 65% of mutations identified. Missense mutations account for more than half of mutations detected; nonsense, deletions, insertions, and splice site mutations have also been reported.73 The frequency of single or several exon deletions/duplications is approximately 10%. A deletion of exon 10 is the most common exonic deletion.88
The few SMAD4 mutations reported thus far in individuals with JP/HHT syndrome are predicted to lead to protein truncation. Although most of the JP/HHT SMAD4 mutations are clustered in exons 10–13, which encodes the MH2 domain, some mutations have also been identified in exons 4–9 encoding the MH1 domain.89 Unlike patients with juvenile polyposis syndrome (JPS), no large SMAD4 deletions/duplications or splice site mutations have been identified to date in patients with JP/HHT.
CLINICAL MOLECULAR ASPECTS
Clinical testing
Molecular genetic testing of ENG, ALK1, and SMAD4 detects mutations in approximately 85% of individuals who meet established clinical diagnostic criteria of HHT.87 Mutations in SMAD4 have been reported in families with a combined syndrome of JPS and HHT.32 Simultaneous testing of ENG and ACVRL1 by both sequence analysis and duplication/deletion analysis is recommended given the relatively high percentage of mutation negative results and uncertain variants by sequence analysis alone. A negative deletion/duplication assay is often helpful in interpreting the sequencing results, in addition to the increased detection it provides.
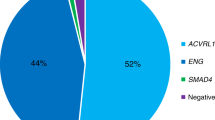
The percentage of mutations in ENG and ACVRL1 is virtually equal (53% and 47%, respectively) after founder effects are excluded.22 The proportion of ENG and ACVRL1 mutations is roughly similar, with a slight preponderance of ENG mutations in North America and in Northern Europe34,90–94 and a greater preponderance of ACVRL1 mutations in Southern Europe.95–97
Sequence analysis of ENG, ACVRL1, and SMAD4 identifies mutations in approximately 75% of individuals with HHT.34,93,94 Sequence variants interpreted to be of uncertain significance are particularly common.
Several techniques including quantitative polymerase chain reaction and multiplex ligation-dependent probe amplification are used to identify deletions not detectable by sequence analysis. The use of one of these methods in addition to sequence analysis increases the detection rate by approximately 10%.34,93
Recent reports suggest that approximately 1–2% of persons clinically diagnosed with HHT will have a mutation detected in the SMAD4 gene or approximately 10% of those who test negative for a mutation in the ENG and ACVRL1 genes.33,34,96 Mutations in SMAD4 have been reported in families with a combined syndrome of JPS and HHT,32 as well as in families reported to have JPS only.98 One study found SMAD4 mutations in 3 of 30 persons who had been referred for DNA-based testing for HHT but were negative for mutations in the ENG and ACVRL1 genes.33 Another study showed that 2% of 194 persons referred for DNA-based testing for HHT had SMAD4 mutations.34 SMAD4 testing is recommended for symptomatic individuals in whom no mutation is identified in ENG or ACVRL1 and in any person with HHT and intestinal polyps.29 There have been four individuals with JPS reported to have missense mutations in ENG.99,100 It is likely that these are not pathogenic mutations. There has been no report of juvenile polyposis in hundreds of patients with HHT reported with ENG mutations.
Testing relatives at risk
Molecular testing should be offered to at-risk, but not clearly affected, relatives if the disease-causing mutation in the family is known, so that morbidity and mortality can be reduced by early diagnosis and treatment. Additionally, relatives who are at genetic risk based on the pedigree can be determined to be free of the family's mutation and thus be spared the anxiety, inconvenience, and cost of repetitive clinical screening. If the disease-causing mutation in the family is not known, it is appropriate to offer clinical diagnostic evaluations to identify those family members who are at risk to be affected and would benefit from early treatment.
Genotype/phenotype
Data suggest that the incidence of certain visceral AVMs depends on the gene mutated, with pulmonary and possibly cerebral AVMs more common in patients with ENG mutations (HHT1) and hepatic AVMs more common in patients with ACVRL1 mutations (HHT2).101–103 However, all these lesions have been seen in individuals with both HHT types.
Pulmonary hypertension in the absence of severe vascular shunting, a rare HHT manifestation, occurs almost exclusively in individuals with mutations in the ACVRL1 gene.104,105
SUMMARY
HHT is underdiagnosed, and affected families are often unaware of the available screening and treatment, resulting in unnecessary life-threatening events in adults and children. The diagnosis typically relies on the presence of characteristic clinical findings in adults, but molecular diagnostics is used to confirm the diagnosis in some cases, to determine the genetic subtype of HHT in a particular family, and to allow for early diagnosis in the youngest generation or two. Optimal medical management for HHT requires distinguishing between organ locations where telangiectases and AVMs are best managed symptomatically/expectantly versus those in which lesions should be detected and treated before the onset of symptoms. International guidelines for the management of HHT have been published as a result of a consensus conference in 2006. This publication reviews the available experience and evidence which lead to the resulting management recommendations.29 In general, telangiectases of the skin, oral and GI mucosa, and liver are treated when symptoms dictate, but AVMs of the lungs and brain are treated in patients without symptoms given their often sudden and catastrophic presentation. This distinction determines routinely recommended screening for AVMs in patients with HHT.
References
Sutton HG . Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Med Mirror 1864; 769–781.
Babington BG . Hereditary epistaxis. Lancet 1865; 2: 362–353.
Marchuk DA, Guttmacher AE, Penner JA, Ganguly P . Report on the workshop on hereditary hemorrhagic telangiectasia, July 10–11, 1997. Am J Med Genet 1998; 76: 269–273.
Guttmacher A, Marchuck D, Pyeritz RE . Hereditary hemorrhagic telangiectasia, 5th ed. Philadelphia, Churchill Livingstone, 2007.
Assar A . The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Am J Gastroenterol 1991; 101: 977–980.
Berg J, Porteous M, Reinhardt D, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet 2003; 40: 585–590.
Plauchu H, de Chadarevian JP, Bideau A, Robert JM . Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32: 291–297.
Porteous ME, Burn J, Proctor SJ . Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet 1992; 29: 527–530.
Morgan T, McDonald J, Anderson C, et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). Pediatrics 2002; 109: E12.
Kjeldsen AD, Kjeldsen J . Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol 2000; 95: 415–418.
Longacre AV, Gross CP, Gallitelli M, Henderson KJ, White RI Jr, Proctor DD . Diagnosis and management of gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol 2003; 98: 59–65.
Ingrosso M, Sabba C, Pisani A, et al. Evidence of small-bowel involvement in hereditary hemorrhagic telangiectasia: a capsule-endoscopic study. Endoscopy 2004; 36: 1074–1079.
Proctor DD, Henderson KJ, Dziura JD, Longacre AV, White RI Jr, Enteroscopic evaluation of the gastrointestinal tract in symptomatic patients with hereditary hemorrhagic telangiectasia. J Clin Gastroenterol 2005; 39: 115–119.
van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ . Real prevalence of pulmonary right-to-left shunt according to genotype in patients with hereditary hemorrhagic telangiectasia: a transthoracic contrast echocardiography study. Chest 2010; 138: 833–839.
Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF . Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am J Respir Crit Care Med 2004; 169: 994–1000.
Gossage JR . Role of contrast echocardiography in screening for pulmonary arteriovenous malformation in patients with hereditary hemorrhagic telangiectasia. Chest 2010; 138: 769–771.
Woods TD, Harmann L, Purath T, et al. Small- and moderate-size right-to-left shunts identified by saline contrast echocardiography are normal and unrelated to migraine headache. Chest 2010; 138: 264–269.
Lovering AT, Stickland MK, Amann M, et al. Hyperoxia prevents exercise-induced intrapulmonary arteriovenous shunt in healthy humans. J Physiol 2008; 586: 4559–4565.
White RI Jr, Pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia: embolotherapy using balloons and coils. Arch Intern Med 1996; 156: 2627–2628.
Shovlin CL, Sodhi V, McCarthy A, Lasjaunias P, Jackson JE, Sheppard MN . Estimates of maternal risks of pregnancy for women with hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): suggested approach for obstetric services. BJOG 2008; 115: 1108–1115.
Cottin V, Dupuis-Girod S, Lesca G, Cordier JF . Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (rendu-osler disease). Respiration 2007; 74: 361–378.
Bayrak-Toydemir P, Mao R, Lewin S, McDonald J . Hereditary hemorrhagic telangiectasia: an overview of diagnostic and management in the molecular era for clinicians. Genet Med 2004; 6: 175–191.
Kjeldsen AD, Oxhoj H, Andersen PE, Green A, Vase P . Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (HHT). J Intern Med 2000; 248: 255–262.
Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G . Hereditary hemorrhagic telangiectasia: multi-detector row helical CT assessment of hepatic involvement. Radiology 2004; 230: 250–259.
Buscarini E, Danesino C, Olivieri C, et al. Doppler ultrasonographic grading of hepatic vascular malformations in hereditary hemorrhagic telangiectasia—results of extensive screening. Ultraschall Med 2004; 25: 348–355.
Buscarini E, Danesino C, Plauchu H, et al. High prevalence of hepatic focal nodular hyperplasia in subjects with hereditary hemorrhagic telangiectasia. Ultrasound Med Biol 2004; 30: 1089–1097.
Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91: 66–67.
Mei-Zahav M, Letarte M, Faughnan ME, Abdalla SA, Cymerman U, MacLusky IB . Symptomatic children with hereditary hemorrhagic telangiectasia: a pediatric center experience. Arch Pediatr Adolesc Med 2006; 160: 596–601.
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. J Med Genet 2011; 48: 73–87.
Curie A, Lesca G, Cottin V, et al. Long-term follow-up in 12 children with pulmonary arteriovenous malformations: confirmation of hereditary hemorrhagic telangiectasia in all cases. J Pediatr 2007; 151: 299–306.
Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 2003; 73: 1240–1249.
Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004; 363: 852–859.
Gallione CJ, Richards JA, Letteboer TG, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet 2006; 43: 793–797.
Prigoda NL, Savas S, Abdalla SA, et al. Hereditary haemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet 2006; 43: 722–728.
Shovlin CL, Letarte M . Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54: 714–729.
Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343: 931–936.
Oxhoj H, Kjeldsen AD, Nielsen G . Screening for pulmonary arteriovenous malformations: contrast echocardiography versus pulse oximetry. Scand Cardiovasc J 2000; 34: 281–285.
Nanthakumar K, Graham AT, Robinson TI, et al. Contrast echocardiography for detection of pulmonary arteriovenous malformations. Am Heart J 2001; 141: 243–246.
Olivieri C, Lanzarini L, Pagella F, et al. Echocardiographic screening discloses increased values of pulmonary artery systolic pressure in 9 of 68 unselected patients affected with hereditary hemorrhagic telangiectasia. Genet Med 2006; 8: 183–190.
Nawaz A, Litt HI, Stavropoulos SW, et al. Digital subtraction pulmonary arteriography versus multidetector CT in the detection of pulmonary arteriovenous malformations. J Vasc Interv Radiol 2008; 19: 1582–1588.
Al-Saleh S, Mei-Zahav M, Faughnan ME, et al. Screening for pulmonary and cerebral arteriovenous malformations in children with hereditary haemorrhagic telangiectasia. Eur Respir J 2009; 34: 875–881.
Faughnan ME, Thabet A, Mei-Zahav M, et al. Pulmonary arteriovenous malformations in children: outcomes of transcatheter embolotherapy. J Pediatr 2004; 145: 826–831.
Ference BA, Shannon TM, White RI Jr, Zawin M, Burdge CM . Life-threatening pulmonary hemorrhage with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Chest 1994; 106: 1387–1390.
Shovlin CL, Winstock AR, Peters AM, Jackson JE, Hughes JM . Medical complications of pregnancy in hereditary haemorrhagic telangiectasia. QJM 1995; 88: 879–887.
Goussous T, Haynes A, Najarian K, Daccarett M, David S . Hereditary hemorrhagic telangiectasia presenting as high output cardiac failure during pregnancy. Cardiol Res Pract 2009; 2009: 437237.
Dupuis-Girod S, Giraud S, Decullier E, et al. Hemorrhagic hereditary telangiectasia (Rendu-Osler disease) and infectious diseases: an underestimated association. Clin Infect Dis 2007; 44: 841–845.
Cahill D, Barker F, Davis K, Kalva S, Sahai I, Frosc M . Case 10-2010: a 37 year old woman with weakness and a mass in the brain. N Engl J Med 2010; 36: 1326–1333.
Gallitelli M, Guastamacchia E, Resta F, Guanti G, Sabba C . Pulmonary arteriovenous malformations, hereditary hemorrhagic telangiectasia, and brain abscess. Respiration 2006; 73: 553–557.
Khalid SK, Pershbacher J, Makan M, Barzilai B, Goodenberger D . Worsening of nose bleeding heralds high cardiac output state in hereditary hemorrhagic telangiectasia. Am J Med 2009; 122: 779.e1–779.e9.
Mahoney EJ, Shapshay SM . New classification of nasal vasculature patterns in hereditary hemorrhagic telangiectasia. Am J Rhinol 2006; 20: 87–90.
Fiorella ML, Ross D, Henderson KJ, White RI Jr, Outcome of septal dermoplasty in patients with hereditary hemorrhagic telangiectasia. Laryngoscope 2005; 115: 301–305.
Hitchings AE, Lennox PA, Lund VJ, Howard DJ . The effect of treatment for epistaxis secondary to hereditary hemorrhagic telangiectasia. Am J Rhinol 2005; 19: 75–78.
Woolford TJ, Loke D, Bateman ND . The use of a nasal obturator in hereditary haemorrhagic telangiectasia: an alternative to Young's procedure. J Laryngol Otol 2002; 116: 455–456.
Jameson JJ, Cave DR . Hormonal and antihormonal therapy for epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 2004; 114: 705–709.
Fernandez LA, Garrido-Martin EM, Sanz-Rodriguez F, et al. Therapeutic action of tranexamic acid in hereditary haemorrhagic telangiectasia (HHT): regulation of ALK-1/endoglin pathway in endothelial cells. Thromb Haemost 2007; 97: 254–262.
Morales-Angulo C, Perez del Molino A, Zarrabeitia R, Fernandez A, Sanz-Rodriguez F, Botella LM . [Treatment of epistaxes in hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease) with tranexamic acid]. Acta Otorrinolaringol Esp 2007; 58: 129–132.
Proctor DD, Henderson KJ, Dziura JD, White RI Jr, Hormonal therapy for the treatment of gastrointestinal bleeding in hereditary hemorrhagic telangiectasia. J Clin Gastroenterol 2008; 42: 756–757.
Flieger D, Hainke S, Fischbach W . Dramatic improvement in hereditary hemorrhagic telangiectasia after treatment with the vascular endothelial growth factor (VEGF) antagonist bevacizumab. Ann Hematol 2006; 85: 631–632.
Trerotola SO, Pyeritz RE . PAVM embolization: an update. Am J Roentgenol 2010; 195: 837–845.
Lee DW, White RI Jr, Egglin TK, et al Embolotherapy of large pulmonary arteriovenous malformations: long-term results. Ann Thorac Surg 1997; 64: 930–939; discussion 939–940.
Moussouttas M, Fayad P, Rosenblatt M, et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology 2000; 55: 959–964.
Post MC, van Gent MW, Snijder RJ, et al. Pulmonary arteriovenous malformations and migraine: a new vision. Respiration 2008; 76: 228–233.
Fulbright RK, Chaloupka JC, Putman CM, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neuroradiol 1998; 19: 477–484.
Garcia-Tsao G . Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol 2007; 46: 499–507.
Wu JS, Saluja S, Garcia-Tsao G, Chong A, Henderson KJ, White RI Jr, Liver involvement in hereditary hemorrhagic telangiectasia: CT and clinical findings do not correlate in symptomatic patients. AJR Am J Roentgenol 2006; 187: W399–W405.
Buscarini E, Plauchu H, Garcia Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int 2006; 26: 1040–1046.
Lerut J, Orlando G, Adam R, et al Liver transplantation for hereditary hemorrhagic telangiectasia: report of the European liver transplant registry. Ann Surg 2006; 244: 854–862; discussion 862–854.
Mitchell A, Adams LA, MacQuillan G, Tibballs J, vanden Driesen R, Delriviere L . Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl 2008; 14: 210–213.
Albinana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabeu C, Botella LM . Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): effects of raloxifene, on endoglin and ALK1 expression in endothelial cells. Thromb Haemost 2010; 103: 525–534.
Bose P, Holter JL, Selby GB . Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 2009; 360: 2143–2144.
Davidson TM, Olitsky SE, Wei JL . Hereditary hemorrhagic telangiectasia/avastin. Laryngoscope 2010; 120: 432–435.
Lebrin F, Srun S, Raymond K, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 2010; 16: 420–428.
Abdalla SA, Letarte M . Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43: 97–110.
Cole SG, Begbie ME, Wallace GM, Shovlin CL . A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet 2005; 42: 577–582.
Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A 2006; 140: 2155–2162.
Rigelsky CM, Jennings C, Lehtonen R, Minai OA, Eng C, Aldred MA . BMPR2 mutation in a patient with pulmonary arterial hypertension and suspected hereditary hemorrhagic telangiectasia. Am J Med Genet A 2008; 146A: 2551–2556.
Oh SP, Seki T, Goss KA, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 2000; 97: 2626–2631.
Li DY, Sorensen LK, Brooke BS, et al. Defective angiogenesis in mice lacking endoglin. Science 1999; 284: 1534–1537.
Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP . Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn 2009; 238: 2479–2493.
Urness LD, Sorensen LK, Li DY . Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 2000; 26: 328–331.
Arthur HM, Ure J, Smith AJ, et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 2000; 217: 42–53.
Bourdeau A, Faughnan ME, Letarte M . Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med 2000; 10: 279–285.
Sabba C, Cirulli A, Rizzi R, et al. Angiogenesis and hereditary hemorrhagic telangiectasia. Rendu-Osler-Weber disease. Acta Haematol 2001; 106: 214–219.
Berg JN, Gallione CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61: 60–67.
Pece N, Vera S, Cymerman U, White RI Jr, Wrana JL, Letarte M . Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest 1997; 100: 2568–2579.
Gallione CJ, Klaus DJ, Yeh EY, et al. Mutation and expression analysis of the endoglin gene in hereditary hemorrhagic telangiectasia reveals null alleles. Hum Mutat 1998; 11: 286–294.
Richards-Yutz J, Grant K, Chao EC, Walther SE, Ganguly A . Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum Genet 2010; 128: 61–77.
McDonald J, Damjanovich K, Millson A, et al. Molecular diagnosis in hereditary hemorrhagic telangiectasia: findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin Genet 2011; 79: 335–344.
Gallione C, Aylsworth AS, Beis J, et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A 2010; 152A: 333–339.
Brusgaard K, Kjeldsen AD, Poulsen L, et al. Mutations in endoglin and in activin receptor-like kinase 1 among Danish patients with hereditary haemorrhagic telangiectasia. Clin Genet 2004; 66: 556–561.
Letteboer TG, Zewald RA, Kamping EJ, et al. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum Genet 2005; 116: 8–16.
Schulte C, Geisthoff U, Lux A, et al. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum Mutat 2005; 25: 595.
Bossler AD, Richards J, George C, Godmilow L, Ganguly A . Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat 2006; 27: 667–675.
Gedge F, McDonald J, Phansalkar A, et al. Clinical and analytic sensitivities in hereditary hemorrhagic telangiectasa testing and a report of de novo mutations. J Mol Diagn 2007; 9: 258–265.
Lenato GM, Lastella P, Di Giacomo MC, et al. DHPLC-based mutation analysis of ENG and ALK-1 genes in HHT Italian population. Hum Mutat 2006; 27: 213–214.
Lesca G, Burnichon N, Raux G, et al. Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Hum Mutat 2006; 27: 598.
Olivieri C, Pagella F, Semino L, et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J Hum Genet 2007; 52: 820–829.
Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet 2004; 41: 484–491.
Sweet K, Willis J, Zhou XP, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA 2005; 294: 2465–2473.
Howe JR, Haidle JL, Lal G, et al. ENG mutations in MADH4/BMPR1A mutation negative patients with juvenile polyposis. Clin Genet 2007; 71: 91–92.
Kjeldsen AD, Moller TR, Brusgaard K, Vase P, Andersen PE . Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med 2005; 258: 349–355.
Bayrak-Toydemir P, McDonald J, Markewitz B, et al. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A 2006; 140: 463–470.
Letteboer TG, Mager JJ, Snijder RJ, et al. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet 2006; 43: 371–377.
Harrison RE, Flanagan JA, Sankelo M, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003; 40: 865–871.
Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345: 325–334.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
McDonald, J., Bayrak-Toydemir, P. & Pyeritz, R. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet Med 13, 607–616 (2011). https://doi.org/10.1097/GIM.0b013e3182136d32
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3182136d32
Keywords
This article is cited by
-
Genetics of brain arteriovenous malformations and cerebral cavernous malformations
Journal of Human Genetics (2023)
-
Imaging to intervention: a review of what the Interventionalist needs to Know about Hereditary Hemorrhagic Telangiectasia
CVIR Endovascular (2021)
-
An advanced ultrasound application used to assess peripheral vascular diseases: superb microvascular imaging
Journal of Echocardiography (2021)
