
Abstract
Purpose: Fragile X syndrome is associated with the expansion of CGG trinucleotide repeats and subsequent methylation of the FMR1 gene. Molecular diagnosis of fragile X currently requires Southern blot analysis to assess methylation. This study describes the evaluation of a polymerase chain reaction-only workflow for the determination of methylation status across a broad range of FMR1 genotypes in male and female specimens.
Methods: We evaluated a novel method that combines allele-specific methylation polymerase chain reaction and capillary electrophoresis with eight cell line and 80 clinical samples, including 39 full mutations. Methylation status was determined using a three-step workflow: (1) differential treatment of genomic DNA using a methylation-sensitive restriction enzyme; (2) polymerase chain reaction with two sets of dye-tagged primers; and (3) amplicon sizing by capillary electrophoresis. All samples were analyzed by both methylation polymerase chain reaction and Southern blot analysis.
Results: FMR1 methylation status and CGG repeat sizing were accurately and reproducibly determined in a set of methylation controls and genomic DNA samples representing a spectrum of CGG repeat lengths and methylation states. Moreover, methylation polymerase chain reaction revealed allele-specific methylation patterns in premutation alleles that were unobtainable using Southern blot analysis.
Conclusions: Methylation polymerase chain reaction enabled high throughput, high resolution, and semiquantitative methylation assessments of FMR1 alleles, as well as determinations of CGG repeat length. Results for all samples were concordant with corresponding Southern blot analyses. As a result, this study presents a polymerase chain reaction-based method for comprehensive FMR1 analysis. In addition, the identification of novel methylation mosaic patterns revealed after polymerase chain reaction and capillary electrophoresis may be relevant to several FMR1 disorders.
Similar content being viewed by others

Main
Fragile X syndrome (FXS, OMIM# 300624) and related disorders are associated with expansion of CGG repeats in the 5′ untranslated region of the fragile X mental retardation-1 (FMR1) gene, OMIM# 309550.1–3 Risk assessment and clinical interpretation of these disorders are defined by the number of CGG repeats and methylation status of the FMR1 gene.4 Expansions in the triplet repeat region to >200 repeats are associated with hypermethylation of the FMR1 gene and the loss of FMR1 protein production, leading to the fragile X phenotype. This phenotype can include mental retardation, autism, and emotional and psychiatric challenges.5 The severity of cognitive disability in FXS patients is not associated with the magnitude of the full-mutation allele6 but does vary with methylation status.7,8 Moreover, premutation alleles (55–200 CGG repeats) are relatively common in the general population occurring in 1 in 130–250 women and in 1 in 250–810 men.9 These allele expansions are associated with two novel disorders: fragile X-associated tremor/ataxia syndrome (OMIM# 300623)10 and fragile X-associated primary ovarian insufficiency (OMIM# 300624).11 Links between the methylation status of premutation alleles and the severity of these disorders have been implicated.12,13 Thus, characterization of repeat length and methylation status for each allele is a critical component of understanding FXS and associated disorders.
FMR1 Southern blot (SB) analysis is currently the gold standard method for the assessment of methylation in expanded alleles. However, this procedure is tedious, low throughput, requires large quantities of genomic DNA (gDNA), and provides inexact sizing across all categories of alleles.4 For these reasons, various polymerase chain reaction (PCR) strategies for assessing methylation status have been reported.14–17 These methods, however, cannot query full-length premutation and full-mutation alleles due to inefficient amplification and/or they fail to provide allele-by-allele resolution of methylation patterns and, thus, are often restricted to analyses of male samples only. To date, no single approach other than SB has demonstrated accurate and comprehensive methylation assessments for expanded alleles in both male and female samples using a robust workflow that is compatible with clinical sample testing.
The lack of established methods that address current limitations in the determination of FMR1 methylation status using SB analysis motivated us to develop a high-throughput, two-color methylation PCR and capillary electrophoresis (CE) procedure (mPCR-CE). This technology leverages the utility of reagents previously demonstrated to reproducibly amplify full-mutation alleles.18 The core approach involves pretreatment of the sample gDNA with HpaII, a methylation-specific restriction digestion enzyme, and, separately, a non-HpaII-treated aliquot of the same DNA, followed by long-range PCR.18 The utility of HpaII in assessing FMR1 methylation using agarose gel electrophoresis was described by Carrel and Willard19 and implemented into a CE workflow for other gene targets.20 We combined HpaII pretreatment and the performance of novel gene-specific FMR1 PCR reagents18 into a two-color PCR workflow that can reproducibly detect all allele repeat lengths, including full mutations with >1000 CGG. This report describes the evaluation of a simple and high-throughput PCR-based FMR1 assay that can accurately assess allele-specific methylation states in both male and female samples, including all premutation and full-mutation alleles, and thus support molecular analyses without the requirement for SB analysis. In addition, the increased sensitivity of mPCR-CE to detect low-abundance alleles, combined with high-resolution CE, revealed unexpected patterns of skewed methylation in female premutation samples that were unresolved using SB analysis.
MATERIALS AND METHODS
Clinical and cell line DNA samples
DNA samples isolated from whole blood were obtained from subjects seen at the M.I.N.D. Institute Clinic after institutional review board approval.18 Cell line DNA samples were obtained from the Coriell Cell Repositories (CCR, Coriell Institute for Medical Research, Camden, NJ). Clinical and cell line DNA samples were diluted to 20 ng/μL in 10 mM Tris, 0.5 mM EDTA, pH 8.8 before PCR.
Preparation of plasmid DNA as CGG DNA, methylation titration, and digestion controls
Plasmid DNA controls containing CGG repeat DNA were prepared from PCR products of FMR1 alleles cloned into pBR322 following standard procedures.21 Five plasmids were constructed: (1) a CGG DNA control to normalize 5′-hexachlorofluorescein (HEX)- and 6′-carboxyfluorescein (FAM)-signal intensities that had a migration window at approximately 40 CGG compared with a clinical sample FMR1 amplicon; (2) a secondary CGG DNA control that contained 90 CGG repeats; (3) a truncated CGG DNA control that contained 44 CGG repeats but an apparent mobility of “0-CGG” on CE analysis; (4) a methylation control used for analytical titration of percent methylation; and (5) a digestion control used in the HpaII treatment reaction to confirm activity of the restriction endonuclease. All plasmids were linearized with HindIII DNA (New England Biolabs, Ipswich, MA).
The primary CGG DNA control was constructed by inserting 30 nucleotides of a nonhuman DNA sequence into a plasmid containing 30 CGG repeats. The CGG DNA control plasmid was prepared at 1.5 × 104 copies/μL. The PCR product of the CGG DNA control migrated with an apparent mobility of approximately 40 CGG repeats (30 CGG + 30 nt of nonhuman DNA). Except where noted, this CGG DNA control was spiked into every PCR and used to normalize signal intensities between the HEX- and FAM-labeled PCR products. In three samples, one FMR1 allele overlapped the CGG DNA control. In these cases, the secondary plasmid DNA, constructed from a 90 CGG DNA PCR product, was used to enable methylation assessment of the sample FMR1 alleles. In a subset of experiments (see Figures, Supplemental Digital Content 1, 2, and 3, http://links.lww.com/GIM/A147, http://links.lww.com/GIM/A148, and http://links.lww.com/GIM/A149), a “truncated CGG” control was constructed to prevent overlap with any FMR1 alleles. This truncated control was prepared from a plasmid that originally contained 44 repeats; however, the common gDNA regions flanking the CGG repeats were excised using standard restriction and ligation procedures to shorten the length of the PCR amplicon. The control retained the FMR1 primer binding sites and produced a PCR product that was 230 bp and had a mobility consistent with a theoretical native FMR1 amplicon without a triplet repeat tract.
Methylation titration controls were prepared from a plasmid containing 30 CGG repeats, cloned from human DNA. An aliquot of this plasmid was methylated with HpaII methyltransferase (New England Biolabs, NEB, Ipswich, MA). Various proportions of the methylated or unmethylated 30 CGG plasmid were admixed to produce 0–100% methylated plasmid DNA. These standard mixtures were diluted to 1.5 × 104 copies/μL in a solution containing 20 ng/μL of cell line DNA (645 CGG repeats, NA04025, CCR). The methylation controls were used to evaluate the analytical recovery of known percent methylation and to assess an in-tube HpaII digestion control. Finally, the digestion control plasmid was premixed with gDNA and included in a supplemental experiment to confirm the elimination of unmethylated FMR1 templates after HpaII digestion.
FMR1 methylation PCR
Two aliquots of 40 ng gDNA (2 μL each of 20 ng/μL) from each sample were prepared for methylation assessment using AmplideX™ Methylation PCR reagents (Asuragen, Inc., Austin, TX). One aliquot was mixed with 4 μL digestion enzyme mix (containing HpaII) and the other with 4 μL digestion control mix (containing digestion buffers). These samples were incubated in separate wells of a sealed 96-well plate at 37°C for 2 hours. After digestion, separate PCR mastermixes of methylation PCR reagents were added to the respective reactions. Each mastermix contained 20 μL mPCR-CE AMP buffer, 0.1 μL GC-rich polymerase mix, 0.4 μL mPCR-CE CGG DNA control (containing 30 CGG + 30 nt nonnative sequence), 2 μL deionized water, and 1.5 μL mPCR-CE primers (forward: 5′TCAGGCGCTCAGCTCCGTTTCGGTTTCA-3′ and reverse: 5′-FAM- or 5′-HEX-AAGCGCCATTGGAGCCCCGCACTTCC). 18 After mixing and mini-spin collection, a 24 μL aliquot of the FAM-primer mastermix was added to the HpaII digested sample, and a 24 μL aliquot of the HEX-primer mastermix was dispensed into the control reaction mixture. PCR was performed with an initial heat denature step of 95°C for 5 minutes, followed by 25 cycles of 97°C for 35 seconds, 62°C for 35 seconds, and 72°C for 4 minutes, and 72°C for 10 minutes. Amplicons were prepared for CE analysis or stored at −15 to −30°C. The sequence of the FMR1 amplicon along with indication of the two HpaII sites that are probed by the mPCR-CE assay is as follows:
5′-TCAGGCGCTCAGCTCCGTTTCGGTTTCA CTT CCGG TGGAGGGCCGCCTCTGAGCGGGCGGCGGGCCGACGGCGAGCGCGGGCGGCGGCGGTGACGGAGGCGCCGCTGCCAGGGGGCGTGCGGCAGCG CGGCGGCGGCGGCGGCGGCGGCGGCGGCGGAGGCGGCGGCGGCGGCGGCGGCGGCGGCGG CTGGGCCTCGAGCGCCCGCAGCCCACCTCTCGGGGGCGGGCTC CCGG CGCTAGCAGGGCTGAAGAGAAGATGGAGGAGCTGGTGGT GGAAGTGCGGGGCTCCAATGGCGCTT-3'
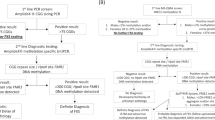
Where the underlined regions designate sequences bound by the forward and reverse primers, the italicized and underlined CCGG sequences indicate the two HpaII sites, and the bolded text represents the CGG repeat region. A schematic of the general workflow, effect of HpaII treatment, and output of CE analysis is shown in Figure 1.

mPCR-CE procedural workflow for the determination of methylation status of FMR1 alleles. Methylation status was assessed by comparing an aliquot of genomic DNA treated with HpaII and amplified with a FAM-labeled primer to an undigested aliquot amplified with a HEX-labeled primer. Unmethylated alleles were digested and, thus, unamplifiable in the subsequent PCR step, whereas methylated alleles were undigested and amplifiable by PCR. A plasmid CGG-rich DNA control was included in each PCR to normalize well-to-well differences in PCR amplification and CE detection efficiency. After PCR, amplicons from both reactions were pooled and injected into a single capillary. In the resulting electropherogram, the CGG repeat length was determined in the HEX channel and the percent methylation computed using a normalized ratio of signals in the FAM channel.
Capillary electrophoresis
A 3130xl Genetic Analyzer (Applied Biosystems Inc. [ABI], Foster City, CA) was used for all experiments except for inter-instrument testing that also used a 3500xL Genetic Analyzer. Two μL of unpurified PCR products (1 μL each from the HEX-labeled products and FAM-labeled products) were mixed with 11 μL of HI-DI Formamide® (ABI) and 2 μL of ROX 1000 Size Ladder (Asuragen), heat denatured at 95°C for 2 minutes, and transferred to the CE system for analysis. Except where noted, all injections followed the standard fragment sizing conditions (36 or 50 cm, POP7) of 2.5 kV for 20 seconds and a 40 minutes run at 15 kV. FMR1 amplicons exceeding 950 bp or approximately 250 CGG repeats had similar mobilities using POP7 on CE and were reported as >250 CGG.18
Data analysis
Electropherograms were analyzed using GeneMapper 4.0 (4.1 for 3500xL data) or PeakScanner V1.0 software (ABI). Peak selection criteria included the following conventions: (1) selection of the highest peak in a single or primary group of amplicons; (2) identification of secondary peak distributions as the first and last peak exceeding 50 rfu; and (3) identification of the tallest peak for alleles detected beyond 820 bp (approximately 200 CGG). We note that secondary peak distributions were predominately observed in female premutation alleles. The conversion of base pair size to number of CGG repeats was determined by linear regression to control amplicons produced from templates with 20, 29, 31, 54, and 119 CGG repeats.18 All product peak sizes for each peak were converted to CGG repeat length using ([sizei−230.2]/2.975). The percent methylation, %Mi, was calculated as a ratio of peak heights between the digested (peaki,FAM) and undigested sample (peaki,HEX) normalized to the CGG DNA control amplicon peak height (CTRLHEX or CTRLFAM) as shown in Equation 1:

Results of CGG repeat length and percent methylation for each detected allele were tabulated in MS Excel. Methylation values that nominally exceeded 100%, as described later, were scored as fully methylated.
SB analysis
SB analysis of the 80 clinical samples was performed as described.22 Cell line DNA was prepared for SB using EcoRI and EagI (NEB) digestion. SB images were assessed categorically (unmethylated, partially methylated, and fully methylated alleles), and the percent of methylation in each sample was determined as reported previously.22,23 Sample results from mPCR-CE and SB were designated as concordant when the percent methylation determined by both methods agreed within 20%. Concordance was also established when the overall methylation status of similarly sized mosaics with different methylation states resolvable by mPCR-CE (e.g., one methylated species and one unmethylated species, or, when taken together, one partially methylated allele) was consistent with unresolved allele methylation state indicated by SB analysis (e.g., a single partially methylated allele). Thus, size mosaics with similar mobilities as resolved by SB analysis but distinguished as separated peaks by mPCR had to be grouped according to the resolution limitations of SB to enable the appropriate method comparisons. Finally, samples that nominally presented >100% methylation were scored as fully methylated for purposes of comparisons with SB analysis.
RESULTS
An essential feature of mPCR-CE is full-length amplification of the entire CGG repeat region, including alleles with >1000 CGG.18 The design of the mPCR-CE methodology is shown in Figure 1. Sample DNA was treated with either HpaII, which degraded the unmethylated templates but retained the methylated alleles, or a control mix, which retained all full-length FMR1 alleles. A CGG-containing PCR control was included in both treatment arms to normalize intertube variation in amplicon yields. A loss in sample amplicon signal on CE indicated digestion and the absence of methylation at either of the two HpaII sites interrogated by the assay, whereas the retention of signal indicated methylation of the corresponding allele at both HpaII sites. The PCR products were then pooled and coinjected into the same capillary. The resulting electropherogram yielded size information for all alleles in the HEX channel and methylation information in the FAM channel. To calculate percent methylation, the ratio of peak heights for each allele was normalized to an external standard that was added to each PCR reaction.
A critical aspect of the mPCR-CE assay design is that two methylation-sensitive HpaII sites are assessed, rather than a single EagI or NruI site that is commonly used for fragile X SB analysis and has been well established to associate genotype and phenotype in patients.23,24 The rationale for an HpaII-based assay design was 2-fold: (1) to ensure continuity with the gene-specific primers used with the corresponding AmplideX™ FMR1 PCR CGG sizing assay18,25 and (2) to minimize the size of the PCR amplicons required to span the CGG repeat region and, thus, retain maximum resolution of the products during CE (as shorter products provide superior separation and more defined peak morphologies compared with longer products). Although previous work demonstrated the reliability of these same two HpaII sites for assessing X-inactivation at the FMR1 locus,19 we pursued an in-depth analytical and clinical sample validation of the mPCR-CE assay to ensure the accurate interpretation of FMR1 methylation data. Consequently, the mPCR-CE assay was evaluated with in vitro methylated DNA standards, cell lines, and clinical samples. Cell line and clinical DNA samples were also analyzed by SB analysis. The purpose of this study was to determine the concordance of mPCR-CE with defined analytical standards, and to compare and contrast mPCR-CE sample assessments with the reference method of SB analysis.
Accuracy, reproducibility, and sensitivity of mPCR-CE with methylated DNA standards and samples
The accuracy of the two-color mPCR-CE workflow was evaluated using a set of quantitative analytical standards containing 30 CGG repeats and known methylation fractions. A total of eight independently quantified standards representing 0–100% methylation were assayed either directly or after the standards were admixed with purified cell line gDNA containing a 645 CGG allele that was known to be fully methylated from SB analysis. A series of electropherograms highlighting the proportional change in signal in the FAM channel for the 30 CGG plasmid control are shown in Figure 2A. As the percent methylation increased, the signal for the 30 CGG template increased relative to the peaks for the CGG DNA control and the admixed 645 CGG full-mutation allele. The change in peak height was normalized to the CGG DNA control peak height and compared with the normalized ratio in the HEX channel (Equation 1). A linear relationship (R2 = 0.998) was observed between the known methylation fraction and the fraction empirically recovered after mPCR-CE (Fig. 2B). Furthermore, the linearity of this relationship was reproducible across two operators on three different run days (R2 = 0.99; data not shown). Methylation of the 645 CGG allele was determined to be quantitative (108% ± 3%) for each of the evaluated standards (Fig. 2B).

Methylation PCR accurately and reproducibly recovers the proportion of methylated DNA standards. A, Electropherograms of PCR products derived from the control (HEX, left) and HpaII-digested (FAM, right) reactions of methylation standards ranging from 0 to 100% methylation are shown. Each reaction included a variable input of methylation standard with a constant concentration of CGG DNA control and 645 CGG full-mutation (FM) allele from cell line DNA (NA04025). Product peaks amplified from the control digestion are shown relative to corresponding percent methylation of the 30 CGG plasmid (30 CGG Std). B, Plot of the linear fit of known input-methylated DNA standards versus detected percent methylation. Quantification of the background, fully methylated 645 CGG allele is superposed (mean = 108% ± 3%) for each methylation standard.
The reproducibility of mPCR-CE was further assessed across eight replicates of two normal alleles (30 and 32 CGG from a female clinical sample) and two full-mutation alleles (550 CGG and 940 CGG) on two CE instrument platforms (an ABI 3130xl and 3500xL). The calculated mPCR-CE methylation fraction yielded a CV ≤8% for the normal alleles and a CV ≤12–16% for the full-mutation alleles across the two different platforms (Table, Supplemental Digital Content 4, http://links.lww.com/GIM/A150). In addition, mPCR-CE results were reproducible across two different operators (CV = 7%) using eight cell line samples, including four full-mutation samples (data not shown).
The sensitivity of mPCR-CE was addressed in two different experiments. In the first, titration of as little as a 1% mass fraction of a clinical full-mutation allele in a background of a normal 31 CGG allele was detectable in both the HEX and FAM channels (Figure, Supplemental Digital Content 1, http://links.lww.com/GIM/A147). In the second, a 10% mass fraction of a fully methylated full-mutation sample (no. 08) could be clearly identified in the background of a 90% mass fraction of a fully unmethylated full-mutation sample (no. 125) (Figure, Supplemental Digital Content 2, http://links.lww.com/GIM/A148). The methylation state of both samples was confirmed by SB analysis (Supplemental Digital Content 5, http://links.lww.com/GIM/A151). Thus, consistent with Figure 2, even a 10% methylation mosaic in the full-mutation range could be detected using clinical samples.
We note that measurements of percent methylation sometimes exceeded 100%. Such values were typically observed within the range of variation of the assay for well-represented alleles (Fig. 2) but were sometimes exaggerated at the lower limits of detection by the CE instrument in methylation assessments of very low-abundance alleles of questionable clinical significance (e.g., inputs of only 1% mass fraction of a clinical full-mutation sample; see Figure, Supplemental Digital Content 1, http://links.lww.com/GIM/A147). This said, it is important to note that methylation assessments in samples with such low allele representation (1% of 80 ng input or 800 pg gDNA equivalent across the two PCR reactions) were still categorically accurate (i.e., the full-mutation allele was correctly interpreted as fully methylated), and the result could be achieved with at least 5000-fold lower input compared with SB.
mPCR-CE assessments of cell line DNA and comparisons with SB analysis
mPCR-CE was evaluated with eight commercially available cell line DNA templates that included normal, premutation, and full-mutation alleles from both male and female samples. Electropherograms of four samples with matching SB data are shown in Figure 3. In each case, the mPCR-CE results were consistent with the corresponding SB data.

Assessments of cell line DNA samples by mPCR-CE and SB across a representative range of genotypes and methylation states. Electropherograms for the control reaction in the HEX channel (left, both methylated and unmethylated templates) compared with electropherograms for the HpaII reaction in the FAM channel (right, methylated templates only). The corresponding SB images for each sample are shown as insets for A, NA06892; B, NA04025; C, NA07537; and D, NA06896. The specific CGG sizes corresponding to bands revealed by SB analysis were mapped from the corresponding mPCR-CE data.
Results for allele size and methylation status from mPCR-CE and SB analyses are summarized in Table 1. The premutation male sample was categorically unmethylated (Fig. 3A), and the four full-mutation samples were categorically fully methylated using both methods. A broad range of full-mutation allele sizes were approximately sized using SB and identified as >250 CGG. Categorically, all male full-mutation alleles were reported as fully methylated by both methods.
In the three female samples, the higher resolution possible by mPCR-CE revealed more nuanced size and methylation mosaicism compared with the lower resolution SB (Table 1). For example, a mosaic female sample (NA06896, “23/95-140” according to CCR) was detected with primary peaks of 23, 112, 136 CGG, and four additional low-abundance peaks of 153, 175, 196, and 219 CGG (Fig. 3D). The 23 CGG and 112 CGG peaks were partially methylated, the 136 CGG peak was fully methylated, and the peaks between 153 and 219 were completely unmethylated. These results were qualitatively similar to the “smear” of detected bands using SB analysis (Fig. 3D). SB revealed a partially unmethylated 23 CGG allele, and a set of expanded unmethylated (<5.2 kb), and methylated alleles. Importantly, mPCR-CE data revealed individual size mosaic contributions to the different allele categories that combined to form the unmethylated smear pattern in SB (Table 1). These initial results with cell line DNA were further substantiated with clinical samples representing a broader number and range of FMR1 genotypes.
Methylation PCR assay performance with clinical DNA samples
A set of 80 clinical specimens representing a range of clinically relevant triplet repeat sizes was evaluated with the FMR1 mPCR-CE assay. The allele sizes for the same samples were determined with gene-specific or CGG repeat primed PCR reported previously.18,25 The cohort of 80 samples comprised 38 male samples (5 normal, 12 premutation, and 21 full mutation) and 42 female samples (9 normal including 1 XXX aneuploidy, 3 intermediate, 18 premutation, and 12 full mutation). mPCR-CE results were obtained for all samples and compared with SB analysis for genotype by size and relative percent methylation. Consistent with the analysis of cell line DNA, mPCR-CE results revealed more detailed molecular information than SB within each genotype category (Table, Supplemental Digital Content 5, http://links.lww.com/GIM/A151).
Identification and resolution of methylation status in full-mutation alleles
The electropherograms and matched SB images for representative full-mutation clinical samples are shown in Figure 4. As described previously,18 the preferred CE configuration used in these experiments provided resolution of CGG repeat length up to approximately 250 CGG. Above this range, full-mutation alleles were detected independent of the repeat length and were identified as >250 CGG. In these cases, the percent methylation represented the aggregate methylation status of the distribution of sizes for the full-mutation allele. As supported by the data below, the overall percentage methylation for the broad category of full mutations was accurately assessed relative to SB analysis. For example, in the male full-mutation sample 88 (Fig. 4A), the expanded allele was detected primarily in the methylated region of the SB with a faint product (approximately 10–20% signal) in the unmethylated region. mPCR-CE revealed 82% methylation for this sample, consistent with the SB result. In the female full-mutation sample (no. 117, Fig. 4B), mPCR-CE results indicated partial (44%) methylation of the 30 CGG allele and full (104%) methylation of the expanded allele consistent with SB analysis for both alleles.

mPCR-CE resolves the full spectrum of methylation status in full-mutation alleles with enhanced sensitivity compared with FMR1 SB. Electropherograms for the control reaction in the HEX channel (left) are compared with electropherograms for the HpaII reaction in the FAM channel (right) for a series of full-mutation samples. The corresponding SB images for each sample are shown as insets for samples A, 88; B, 117; C, 125; and D, 09.
mPCR-CE simplified the identification of methylation status in more complex samples that can be problematic for SB analysis. The male full-mutation sample 125 (Fig. 4C) presented a very low-intensity smear across the unmethylated region (3 to >5 kb). Using mPCR-CE, the full-mutation peak was clearly detected in the HEX but not the FAM channel despite robust amplification of the corresponding reference CGG DNA PCR control in both channels. Thus, the full-mutation allele was unmethylated. In the final example (Fig. 4D), an unusual pattern of size and methylation mosaicism was resolved. By SB analysis, the full-mutation allele appeared partially methylated, whereas mPCR-CE revealed two distinct allele sizes with skewed methylation. The predominant full-mutation signal was observed in the unmethylated longer template(s), whereas the less abundant templates at approximately 200 CGG were fully methylated. Thus, the differential methylation of theses two size mosaics was more clearly resolved by mPCR-CE, and the methylation pattern was more complex than that indicated by SB.
Identification of novel skewing patterns in female premutation alleles that were undetected by SB analysis
Electropherograms and matched SB images for four representative female premutation samples are shown in Figure 5. The characteristic feature of each sample was the detection of two groups of repeat sizes for the longer allele, namely 53 and 54–56 CGG; 70 and 71–75 CGG; 90 and 92–101 CGG; and 107 and 109–125 CGG (Fig. 5A–D, respectively). After digestion and amplification with FAM-labeled primers, only the shorter CGG repeat lengths within each mosaic sample were detected. The size mosaicism was neither observed in the HpaII-treated FAM channel of any female samples nor was it observed after the PCR of any of the 12 tested male premutation alleles of comparable repeat length. Thus, we conclude that this pattern was not an artifact of polymerase “stuttering” but a true representation of the biological heterogeneity of the female premutation allele.

mPCR-CE identifies novel patterns of skewed methylation in female premutation alleles that are obscured using low-resolution SB. Electropherograms for the control reaction in the HEX channel (left) are compared with electropherograms for the HpaII reaction in the FAM channel (right) for a series of female premutation samples. In each case, differential and skewed methylation patterns were observed (plot insets). CGG repeat lengths were reported as the highest peak in the primary group of alleles and as a range of peaks exceeding 50 rfu for the secondary group. The corresponding SB images are included for samples A, 72; B, 95; C, 75; and D, 76.
In these samples with two populations of allele sizes in the premutation allele, the more expanded CGG repeats were completely unmethylated. This unexpected size mosaicism and skewed methylation pattern was uniquely identified in 83% (15/18) female premutation samples within the tested cohort. In contrast, the resolution limitations of SB yielded only one detectable premutation band in each case and, thus, masked this level of biological detail. We also note that the skewed mosaicism observed was not specific to an analysis of methylated HpaII sites. In fact, the same pattern was observed when the template was treated with EagI, rather than HpaII, and analyzed using a modification of the mPCR-CE method that permitted the assessment of EagI methylated sites (Figure, Supplemental Digital Content 3, http://links.lww.com/GIM/A149).
Concordance with SB analysis
The CGG repeat length and percent methylation were tabulated for each clinical sample and compared with the results of previously determined SB analysis (Table, Supplemental Digital Content 5, http://links.lww.com/GIM/A151). Because of the higher resolution of CE, some alleles that were readily distinguished by mPCR-CE were unresolved by SB (such as two normal premutation alleles of similar size). In these cases, alleles were grouped to match the resolution limits of SB analysis. For example, sample 75 (Fig. 5C) revealed two allele populations of 90 CGG (99% methylation) and 92-101 CGG (0% methylation) with an overall grouped “partial methylation” status for that allele. This grouping was consistent with SB analysis for the unresolved migration difference between these two size mosaics and an overall 50% methylation status (Table, Supplemental Digital Content 5, http://links.lww.com/GIM/A151). In another similar case, sample 09 (Fig. 4D) presented two full-mutation allele populations with distinct methylation status. These alleles were aggregated to match the sizing ambiguity of SB and combined for partial methylation of the expanded allele. The results for all samples having at least one allele ≥55 repeats, as determined using mPCR-CE, are summarized in Table 2 within genotype and methylation categories. As expected, full-mutation alleles were typically methylated, and premutation alleles were unmethylated. Across 63 samples with expanded alleles, including 33 full mutations, the mPCR-CE results were in perfect agreement with SB analysis.
DISCUSSION
Determination of FMR1 CGG repeat size and methylation status currently requires a combination of PCR and SB analysis.4 SB analysis is a labor-intensive process that provides an approximation of triplet repeat length and a visual estimate of methylation for only those alleles that can be well resolved by slab gel electrophoresis. This sizing limitation reduces the ability to distinguish large premutation from full-mutation alleles and can cause large uncertainties in sizing accuracy. PCR in combination with CE offers sizing accuracy within 1 or a few triplet repeats,25,26 but technical challenges in the amplification of large CGG expansions have historically restricted repeat quantification to normal and smaller premutation alleles. Nevertheless, the strong need to improve the workflow and data quality from SB analysis has motivated a number of studies that have attempted to assess FMR1 methylation status by alternative methods.14–17,27–31
Most of the described methods have exploited the resistance of 5-methylcytosine to bisulfite conversion to reveal the corresponding epigenetic mark. When appropriate conversion procedures and controls are used, bisulfite methods are accurate and highly reproducible.32,33 For FMR1, however, bisulfite-based methylation PCR methods have been practically limited to evaluations of male samples only (because of the mixed methylation states that confound interpretations of female samples) and/or have demonstrated limited utility for expanded alleles. In general, published methods have required the use of primers that are complementary to the repeat region to create a series of amplicons of various lengths that telegraph the presence of an expanded allele29 or have relied on short amplicons that flank representative methylation sites as an alternative to amplifying the entire CGG repeat region.28 For example, Zhou et al.29 modified the conventional bisulfite approach using a three-color, three-PCR methodology compatible with CE analysis. Although the method did identify methylation status in full-mutation alleles, large female premutation alleles confounded the results, and underlying allele-specific methylation patterns were excluded. In addition, Coffee et al.30 used a quantitative methylation-sensitive PCR to identify FXS males, but not females, in a pilot newborn screening study. As an example of alternatives to bisulfite treatment, Nygren et al.28 described a multiplex ligation probe amplification procedure of endonuclease-treated DNA, but, again, the method was only suitable for the analysis of male samples.
In this study, we evaluated a simple PCR and CE workflow to accurately determine the allele size and specific methylation state of normal and expanded FMR1 alleles in both male and female samples. Methylated and unmethylated alleles were differentiated using a methylation-sensitive restriction enzyme, HpaII, and color-specific PCR primers. We note that the utility of HpaII digestion in assessing FMR1 methylation, and the activity of the X chromosome in general, was previously established by Carrel and Willard.19 A key innovation that enabled the analysis of methylation status by mPCR-CE was the development of a PCR assay for the analysis of full-mutation FMR1 alleles.18 In addition, plasmid controls that contain CGG repeats offered improved normalization of signal intensities and supported robust quantification of the methylation fraction for each allele.
The mPCR-CE method provided a streamlined workflow by eliminating purification steps and minimizing reagent transfer steps. The procedure can support high-volume sample analyses, requiring approximately 2 hours of total hands-on time (much of which can be automated) to process up to 48 samples per operator. Another advantage of the method is that mPCR-CE requires an input of only 80 ng of DNA, which is 50- to 100-fold less than SB and 10-fold less than bisulfite15,27,29 or other HpaII-mediated PCR methods.19,34,35
mPCR-CE yielded concordant and novel information with respect to SB analysis. In both methods, a methylation-sensitive restriction enzyme that fails to cleave methylated sites is used to delineate methylated and nonmethylated alleles. We compared HpaII-digested DNA followed by PCR-CE with SB analysis and demonstrated concordance between the two methods. We also note that the data outcomes relevant for FMR1 analysis are the CGG repeat length and the allele-specific methylation fraction. This fraction is the reciprocal of the FMR1 activation ratio for normal alleles.36 Importantly, mPCR-CE determination of the X-activation ratio from 8 normal females (0.51) and 12 female full mutations (0.67) in our study (Table, Supplemental Digital Content 5, http://links.lww.com/GIM/A151) was consistent with previously published data derived from SB analysis of EagI-digested gDNA.6
A novel outcome of this study was the finding that high-resolution methylation assessments of the FMR1 repeat region revealed molecular information that was undetectable in SB images. For example, mPCR-CE identified low-abundance mosaic alleles (Figs. 2 and 4 and Supplemental Digital Content 1 and 2, http://links.lww.com/GIM/A147 and http://links.lww.com/GIM/A148) and indicated differential methylation of premutation and full-mutation alleles (Figs. 4 and 5). mPCR-CE also revealed novel skewing patterns in 15 of 18 female premutation alleles (Fig. 5 and Table, Supplemental Digital Content 5, http://links.lww.com/GIM/A151). These skewed methylation patterns were observed irrespective of whether the methylation site interrogated by the mPCR-CE reagents was HpaII or EagI (Figure, Supplemental Digital Content 3, http://links.lww.com/GIM/A149). The implications of these findings are the subject of ongoing experiments, but we speculate that such skewed methylation may be linked to the instability of the premutation allele.
Potential limitations of the mPCR-CE technique include overestimation of methylation resulting from incomplete HpaII digestion, lack of resolution for full-mutation sizing beyond 250 CGG, and, infrequently, comigration of the CGG repeat control with amplicons of biological alleles of similar size. Each of these limitations can be addressed. Evidence of incomplete digestion was not observed in our study. Even so, a spiked-in digestion control would be valuable in routine mPCR-CE assays to assess the efficiency of digestion of each sample rather than rely on digestion controls that are run with each batch of samples. In preliminary testing, a plasmid that contained unmethylated HpaII sites was mixed with gDNA before HpaII treatment or the control reaction. This digestion control eliminated 97% of the plasmid amplicon signal (Table, Supplemental Digital Content 6, http://links.lww.com/GIM/A152). Sizing and methylation assessment of a fully methylated full-mutation allele and an added analytical standard with known 20% methylation recovered the expected methylation fractions and were unaffected by addition of the digestion control. These results suggest the compatibility of such a control in future studies.
Furthermore, constraints of CE that limit the sizing window for FMR1 amplicons to approximately 250 CGG can be expanded using agarose gel electrophoresis, as demonstrated in our previously published report.18 Amplicons produced from the control digestion reaction can be separated by agarose gel, and the CGG repeat length can be determined relative to an appropriate DNA ladder. Thus, this simple step can circumvent the need for a cumbersome SB process to size very long expanded alleles.
Finally, we note that 3 of 80 (3.75%) samples in our clinical sample cohort presented at least one allele amplicon that overlapped with the plasmid control amplicon detected at approximately 40 CGG after CE. The methylation status of these three samples was determined using an alternative plasmid control (a pBR322 construct from a 90 CGG amplicon). We are currently evaluating a GC-rich control that migrates outside of the biologically relevant repeat region to address this issue. This control includes a series of CGG repeats but migrates as a PCR product with “0-CGG” due to the deletion of sequences outside of the CGG repeat region. The 0-CGG control was successfully used in preliminary studies as the normalizing signal amplicon in Figures, Supplemental Digital Content 1 and 2, http://links.lww.com/GIM/A147 and http://links.lww.com/GIM/A148. The possible range of CGG repeat lengths in the normal region suggests that implementation of this truncated DNA control will facilitate routine FMR1 characterization without the need to accommodate overlap with target alleles.
To date, the mPCR-CE technology evaluated in this study is the only PCR-based methodology that has been reported to detect and resolve methylation status from full-length triplet-repeat-containing amplicons in both male and female samples with concordant results to SB analysis. The procedure is amenable to routine analyses and high-throughput screening applications. The implementation of mPCR-CE may depend on the needs of the laboratory environment. In one possible workflow, our previously described repeat primed PCR could be used for assessment of size, zygosity, and indication of AGG interrupting sequences for all incoming samples.25 Reflex methylation assessments could be performed using mPCR-CE for only those premutation and full-mutation samples that require such analyses. This strategy may be particularly effective for large sample sets of diverse allele genotypes that require comprehensive molecular characterization. Alternatively, mPCR-CE could be used to analyze all samples, such that CGG repeat length and methylation status would be obtained in a single PCR workflow without the complications of reflex testing.
In conclusion, we propose that mPCR-CE offers a technology foundation for comprehensive FMR1 molecular assessments using a PCR-only assay approach. Moreover, this approach has the potential to provide new molecular information unresolvable by SB analysis that may support a more complete understanding of fragile X disorders.
References
Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Oberle I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991; 252: 1097–1102.
Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991; 65: 905–914.
Sherman S, Pletcher BA, Driscoll DA . Fragile X syndrome: diagnostic and carrier testing. Genet Med 2005; 7: 584–587.
Hagerman RJ, Hagerman PJ . Testing for fragile X gene mutations throughout the life span. JAMA 2008; 300: 2419–2421.
de Vries BB, Wiegers AM, Smits AP, et al. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet 1996; 58: 1025–1032.
Wohrle D, Salat U, Glaser D, et al. Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J Med Genet 1998; 35: 103–111.
Smeets HJ, Smits AP, Verheij CE, et al. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet 1995; 4: 2103–2108.
Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004; 291: 460–469.
Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001; 57: 127–130.
Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study—preliminary data. Am J Med Genet 1999; 83: 322–325.
Bodega B, Bione S, Dalpra L, et al. Influence of intermediate and uninterrupted FMR1 CGG expansions in premature ovarian failure manifestation. Hum Reprod 2006; 21: 952–957.
Berry-Kravis E, Potanos K, Weinberg D, Zhou L, Goetz CG . Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann Neurol 2005; 57: 144–147.
Zhou Y, Law HY, Boehm CD, et al. Robust fragile X (CGG)n genotype classification using a methylation specific triple PCR assay. J Med Genet 2004; 41: e45.
Panagopoulos I, Lassen C, Kristoffersson U, Aman P . A methylation PCR approach for detection of fragile X syndrome. Hum Mutat 1999; 14: 71–79.
Das S, Kubota T, Song M, et al. Methylation analysis of the fragile X syndrome by PCR. Genet Test 1997; 1: 151–155.
Weinhausel A, Haas OA . Evaluation of the fragile X (FRAXA) syndrome with methylation-sensitive PCR. Hum Genet 2001; 108: 450–458.
Filipovic-Sadic S, Sah S, Chen L, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem 2010; 56: 399–408.
Carrel L, Willard HF . An assay for X inactivation based on differential methylation at the fragile X locus, FMR1. Am J Med Genet 1996; 64: 27–30.
Beever CL, Stephenson MD, Penaherrera MS, et al. Skewed X-chromosome inactivation is associated with trisomy in women ascertained on the basis of recurrent spontaneous abortion or chromosomally abnormal pregnancies. Am J Hum Genet 2003; 72: 399–407.
Sambrook J, Fritsch EF, Maniatis T . Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor, NY, CSHL Press, 1989.
Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ . A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn 2008; 10: 43–49.
Taylor AK, Safanda JF, Fall MZ, et al. Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA 1994; 271: 507–514.
Rousseau F, Heitz D, Biancalana V, et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med 1991; 325: 1673–1681.
Chen L, Hadd A, Sah S, et al. An information-rich CGG repeat primed PCR assay that detects the full range of expanded alleles and minimizes the need for Southern blotting in FMR1 analysis. J Mol Diagn 2010; 12: 589–600.
Amos Wilson J, Pratt VM, Phansalkar A, et al. Consensus characterization of 16 FMR1 reference materials: a consortium study. J Mol Diagn 2008; 10: 2–12.
Dahl C, Guldberg P . A ligation assay for multiplex analysis of CpG methylation using bisulfite-treated DNA. Nucleic Acids Res 2007; 35: e144.
Nygren AO, Lens SI, Carvalho R . Methylation-specific multiplex ligation-dependent probe amplification enables a rapid and reliable distinction between male FMR1 premutation and full-mutation alleles. J Mol Diagn 2008; 10: 496–501.
Zhou Y, Lum JM, Yeo GH, Kiing J, Tay SK, Chong SS . Simplified molecular diagnosis of fragile X syndrome by fluorescent methylation-specific PCR and GeneScan analysis. Clin Chem 2006; 52: 1492–1500.
Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009; 85: 503–514.
Tzeng CC, Liou CP, Li CF, et al. Methyl-CpG-binding PCR of bloodspots for confirmation of fragile X syndrome in males. J Biomed Biotechnol 2009; 2009: 643–692.
Ogino S, Kawasaki T, Brahmandam M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn 2006; 8: 209–217.
Genereux DP, Johnson WC, Burden AF, Stoger R, Laird CD . Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies. Nucleic Acids Res 2008; 36: e150.
Kline J, Kinney A, Levin B, et al. X-chromosome inactivation and ovarian age during the reproductive years. Fertil Steril 2006; 85: 1488–1495.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW . Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Rousseau F, Heitz D, Oberle I, Mandel JL . Selection in blood cells from female carriers of the fragile X syndrome: inverse correlation between age and proportion of active X chromosomes carrying the full mutation. J Med Genet 1991; 28: 830–836.
Acknowledgements
This project was supported, in part, by awards from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, including R43HD060450 and R44HD060450 to A.G.H., HD02274 to F.T., and HD040661 to P.J.H. The authors acknowledge Dr. Timothy Stenzel and Dr. Stephen Brown for helpful discussions.
Disclosure
L.C., A.G.H., S.S., J.F.H., S.F-S. and G.J.L. are employees of Asuragen, Inc. and hold stock or stock options in the company.
Consultant or Advisory Role: P.J.H., National Fragile X Foundation (self and immediate family member), Asuragen, Novartis (immediate family member), and Roche (immediate family member).
Stock Ownership: A.H., S. S., L.C., J.H., S.F-S. and G.J.L., Asuragen.
Honoraria: P.J.H., 2008 4th Annual Advances in Neurology Conference (Houston, TX), 2009 McMaster University external thesis reader, 2008 Muscular Dystrophy Society international meeting (faculty stipend), Elsevier Journal of Molecular Biology editorial board (manuscript-handling fees), University of California San Francisco talk (March 2009), University of California San Francisco talk (2009, immediate family member), Institute for Basic Research talk (Staten Island, NY; immediate family member), and University of North Carolina talk (2009, immediate family member).
Research Funding: P.J.H., Neuropharm (immediate family member), Seaside Therapeutics (immediate family member), Coleman Institute (immediate family member), National Fragile X Foundation (self and immediate family member), and NIH (self and immediate family member). This project was supported in part by awards from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, including R43HD060450 and R44HD060450 to A.G.H., HD044410 to F.T., and HD040661 to P.J.H.
Expert Testimony: P.J.H., Mullin and Obstetrix case (fragile X testing, immediate family member).
Other Remuneration: P.J.H., Seaside Therapeutics (immediate family member), Roche (immediate family member), Novartis (immediate family member), New Orleans Human Development Center Institute, Movement Disorder Society 12th International Congress, Colorado Clinical Scholars Seminar, Nanomedicine Workshop, Nanomedicine Retreat, Washington University Brain Awareness Week, XLMR Meeting (Brazil), and University of California San Francisco Memory and Aging Center.
Patent Pending: P.J.H. and F.T. (Method for detection of expanded CGG repeats; U.S. Patent Application No.: 11/779,873).
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally to the work.
Disclosure: See Acknowledgments for author disclosures.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
Chen, L., Hadd, A., Sah, S. et al. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet Med 13, 528–538 (2011). https://doi.org/10.1097/GIM.0b013e31820a780f
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31820a780f
Keywords
This article is cited by
-
The Effects of Dual Task Cognitive Interference and Fast-Paced Walking on Gait, Turns, and Falls in Men and Women with FXTAS
The Cerebellum (2021)
-
Abnormally Methylated FMR1 in Absence of a Detectable Full Mutation in a U.S.A Patient Cohort Referred for Fragile X Testing
Scientific Reports (2019)
-
Use of human-derived stem cells to create a novel, in vitro model designed to explore FMR1 CGG repeat instability amongst female premutation carriers
Journal of Assisted Reproduction and Genetics (2018)
-
Association of skewed X-chromosome inactivation with FMR1 CGG repeat length and anti-Mullerian hormone levels: a cohort study
Reproductive Biology and Endocrinology (2017)
-
Fragile X syndrome: a review of clinical and molecular diagnoses
Italian Journal of Pediatrics (2017)
