
Abstract
Purpose: To determine the frequency of SMN1 deletion carriers in the Israeli population and to assess the feasibility of population screening for spinal muscular atrophy.
Methods: A total of 6394 individuals without family history of spinal muscular atrophy underwent genetic screening by multiplex ligation-dependent probe amplification, designed to detect SMN1 exon 7 and exon 8 copy number.
Results: One hundred fifty-nine individuals carried an SMN1 heterozygous exon 7 deletion, yielding a carrier frequency of 1:40. About 10.8% of individuals were found to carry two or more SMN1 exon 7 copies on the same chromosome (cis configuration). This implies that some deletion carriers may not be detected by multiplex ligation-dependent probe amplification or similar quantitative methods. The acceptance of spinal muscular atrophy screening among women undergoing testing for fragile X syndrome and cystic fibrosis reached 93%.
Conclusions: Currently used molecular techniques cannot detect about 5% of spinal muscular atrophy carriers with a cis configuration or individuals with SMN1 sequence mutations and de novo deletions. Thus, it is estimated that the spinal muscular atrophy carrier detection rate is about 90%. Given the severity of spinal muscular atrophy, the relatively high carrier frequency, and the estimated detection rate, we conclude that population-based screening for spinal muscular atrophy is feasible and acceptable.
Similar content being viewed by others


Main
Spinal muscular atrophy (SMA; type I, OMIM# 253300; type II, OMIM# 253550; type III, OMIM# 253400; and type IV, OMIM# 271150)1 is a relatively common neuromuscular genetic disorder caused by the degeneration of motor neurons in the anterior horn of the spinal cord. The degenerative process leads to progressive muscular atrophy, which, in the most severe form, causes respiratory failure and infantile death. SMA is considered to be the second most common fatal childhood autosomal recessive disease.2 The disease is categorized into four types (I–IV), based on age of onset and severity, with SMA Type I (Werdnig-Hoffmann disease) being the most common and severe form, usually resulting in death by the age of 2 years.
SMA is prevalent worldwide, with an estimated incidence of 1:6,000–1:10,000 and a carrier frequency of 1/38–1/50.3,4 About 93% of childhood patients with SMA have a homozygous deletion of exon 7 of the survival motor neuron 1 (SMN1) gene (genotype 0/0). The remaining patients carry a deletion on one allele and an intragenic mutation on the other allele (0/SMN1m genotype).5,6 Occurrence of the disease as a result of two intragenic mutations is extremely rare.
Given the severity of this disease, the relatively high carrier frequency rate, and the current lack of effective treatment, population-based carrier screening for SMA may be justified. Indeed, the American College of Medical Genetics recently recommended that genetic SMA carrier screening should be made available to all US couples.7 Conversely, the American College of Obstetricians and Gynecologists stated in their committee opinion8 that “preconception and prenatal screening for SMA is not recommended in the general population at this time.” They stated that several issues including critical assessment of pilot screening programs, cost-effectiveness analysis, development of appropriate educational materials, and development of laboratory essay standards are required before using panethnic prenatal screening for SMA.
Detecting heterozygous SMN1 deletion carriers is technically difficult because of the presence of a highly homologous gene SMN2. The SMN1 and SMN2 genes are distinguished by a small number of polymorphisms, five of them map to exons and none of them is translationally significant.5,9 The most important of these, a C>T base change maps to exon 7 and confers an alternative splicing of SMN2 exon 7,10 resulting in a modified SMN protein with both limited half-life and function. Determination of SMN1 exon 7 copy number has been previously reported using one of several polymerase chain reaction-based dosage assays3,6,11,12 or by multiplex ligation-dependent probe amplification (MLPA).13–15
Carrier screening for SMA has several inherent limitations: (1) Some heterozygous deletion carriers have two copies of the SMN1 gene on the same chromosome (cis configuration or 2/0 genotype). Thus, any quantitative analysis of SMN1 copy number would not differentiate these carriers from normal individuals having two SMN1 copies, one on each chromosome (trans configuration or 1/1 genotype).11,16 (2) There is a relatively high rate of de novo mutations in the SMN1 gene. These mutations have been estimated to account for about 2% of cases.17 (3) Rare SMN1 intragenic mutations, when paired with the far more common SMN1 deletion may be responsible for SMA but are nonetheless undetectable by dosage analyses.5,6
To date, a few population-based studies for SMA have been performed,4,18–20 most were relatively limited in size and were not performed as a part of a comprehensive population-based carrier screening program. Interestingly, a recent study by Hendrickson et al.21 studied differences in SMA carrier frequencies among several ethnic groups within North America. They reported a carrier rate of 1:46 among Ashkenazi Jews and 1:37 among other Caucasians.21 In addition, they noted a significantly lower carrier rates among African Americans (1:91) and Hispanics (1:125). A recent study of SMA screening in Israel reported a much lower carrier rate of 1:62, with no statistically significant differences among ethnic groups.22 Interestingly, they detected a significantly higher rate of SMN1 exon 7 duplication among Ashkenazi Jews (13.5%) compared with only 6% among North African Jews. They suggested that this may impact on a different rate of false-negative results among different ethnic groups.
The objective of our study was to describe our experience with the addition of SMA testing to the population-based genetic screening program that has long been implemented in Israel.23 In addition, because the occurrence of two or more SMN1 exon 7 copies in cis impacts detection rates, we sought to determine the frequency of such genotypes in the Israeli population and evaluate their impact on prenatal diagnosis and genetic counseling.
MATERIALS AND METHODS
Patients
Testing for SMA carriers has been recommended by the Israeli Society of Medical Genetics since March 2007. The study included 6394 consecutive individuals from the general population, who requested SMA carrier testing between March 2007 and August 2009. Each individual was required to fill in a routine questionnaire regarding family history to ensure the absence of positive family history of SMA among the screened population. The study was approved by the Institutional Review Board (TLV0549-09).
Multiplex ligation-dependent probe amplification
DNA was extracted from blood samples using routine salting out methods. The number of SMN1 gene copy number was estimated based on the analysis of SMN1 exons 7 and 8.
The MLPA technique was used according to the manufacturer's instructions (MRC-Holland, Amsterdam, the Netherlands). The assay was carried out using the SALSA® MLPA® kit P021 in a thermal cycler. The kit contains multiple amplification probes on chromosome 5q, in and around the SMN1 gene locus. Of these, the most important probes allow the determination of SMN1 and SMN2 exon 7 and 8 as well as that of the adjacent gene BIRC1 (NAIP). The SMN exon 7 probes 1260-L0966 and 1260-L0967 have a ligation site at the C→T transition locus: the SMN1 sequence has a C at the ligation site (resulting in a 270 nt peak), whereas the SMN2 sequence has a T at the ligation site (resulting in a 276 nt peak). Therefore, this probe allows copy number estimation for both SMN1 and SMN2. Similarly, the SMN exon 8 probes 1812-L1372 and 1812-L1373 distinguish between SMN1 and SMN2 at the G→A transition site. In addition, two probes were located within the BIRC1 gene (238 nt and 346 nt). Several control probes mapped to other autosomes.
After the MLPA assay, aliquots of the products were analyzed on an ABI Genetic Analyzer (ABI 310, 3100, or 3130xl; Applied Biosystems, USA) with LIZ500 (Applied Biosystems, Foster City, CA) as the internal size control. The chromatogram of each sample was then analyzed by ABI software (GeneMapper version 4.0 or GeneScan 3.7) followed by the Coffalyzer software supplied by the manufacturer (MRC-Holland). This Microsoft Excel-based program normalizes the data, taking into account the characteristic effect of most MLPA probe mixes, namely, the probe-length-dependent decrease in peak area/height. This software calculates the relative peak area (RPA) for each probe in the tested sample and compares it with the RPA in normal controls, known to have two SMN1 copies (defined as RPA = 1). The ratios were presented in a tabular form. The genotypes were then classified according to cutoff values. The following cutoff values were used, as suggested by the distributor: a ratio of 0.3–0.7 indicating the presence of a single copy, a ratio of 0.71–1.35 indicating two copies, and a ratio >1.35 indicating the presence of three or more copies. Cases detected as having a single copy or borderline values were confirmed by repeat analyses.
RESULTS
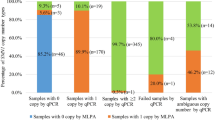
One hundred fifty-nine of the 6394 healthy individuals were found to be SMA carriers, yielding a carrier frequency of 1:40 (∼2.5%, Table 1) with an allele frequency of 1:80.
Furthermore, in 128 individuals with SMN1 exon 7 deletion (80%), the deletion also spanned SMN1 exon 8. Deletions that included both SMN1 exons 7 and 8 and the adjacent BIRC1 gene were observed in 52% (82/159) of cases, indicating an unusually high level of copy number variation within this genomic region.
Homozygous deletions of SMN1 exon 8 alone were detected in two healthy individuals. Homozygous deletions of BIRC1 alone were found in 10 healthy individuals.
Three or more copies of SMN1 exon 7 were detected in 690 individuals (10.8%, Table 1). Assuming that the majority of individuals with three or more SMN1 copies have at least one copy on each chromosome, the estimated allele frequency of the cis configuration is ∼1:19 (5.5%). Therefore, about 1:760 individuals having two SMN1 exon 7 copies may still be deletion carriers (2/0 genotype). This fact highlights the limitation of all quantitative SMN1 analyses that underestimate the SMA carrier rate.2
Copy number analysis using MLPA requires transformation of a continuous variable (RPA, as generated by the Coffalyzer algorithm) into an integer (corresponding to gene copy number). As expected, the computed distribution of the RPA values for SMN1 exon 7 demonstrates a trimodal, bell-shaped distribution curve, around the values of 0.5, 1.0, and 1.5 (corresponding to one, two, and three copies, respectively; Fig. 1). However, appropriate cutoffs must be determined if this test is to be used in a clinical setting. Unfortunately, there is no clear demarcation between the three peaks, and no clear cutoff values exist. The use of arbitrarily determined cutoffs will generate false-positive and/or false-negative results. For example, changing the cutoff value between one and two SMN1 exon 7 copies from 0.7 to 0.75 will increase the number of individuals determined as carriers by 8% (12 individuals, Table 2). Interestingly, although more than 80% of individuals with the 1:0 genotype, as determined by the 0.7 cutoff, have a single copy of exon 8, only 2% of individuals with a value of 0.71–0.8 have a single copy of exon 8 (Table 2). This suggests that the majority of individuals in this range are not true carriers. The cutoff value between two and three SMN1 copies is even less obvious, with a greater degree of overlap. However, this usually has a little clinical significance.

Distribution of SMN1 exon 7 copy number, using the MLPA method. The total number of 6394 screened individuals (y-axis) plotted against increasing RPA values (x-axis). Note that although the highest number of individuals are distributed around RPA levels of 0.5, 1.0, and 1.5 representing one, two, and three copy number of SMN1 exon 7, respectively, it is impossible to unequivocally identify cutoff values that distinguish the three regions.
All SMA carriers detected by our program were given detailed genetic counseling and were made aware of the disease characteristics, inheritance pattern, and reproductive options. Carrier testing was recommended to the partners of all carriers. (It should be noted that some partners underwent SMA carrier testing at other institutions as required by their medical insurance coverage.) During the study period, a total of 65 partners of SMA carriers detected by our program underwent SMA carrier testing in our laboratory. Of these, none were found to be SMA carriers.
Prenatal diagnosis for SMA was performed for six cases detected by our screening program. In two couples, the partners of the SMA carriers detected by our program were detected as SMA carriers by other laboratories. Prenatal diagnosis performed by amniocentesis revealed that in both cases, the fetuses were unaffected. In another case, an SMA carrier mother underwent prenatal diagnosis by amniocentesis because her partner was unavailable for testing. The fetus was unaffected. In two more cases, molecular testing for SMA was performed on the fetuses of SMA carrier mothers whose partners were SMA screen-negative. In both cases, invasive prenatal diagnosis was performed for other indications. Both fetuses were unaffected. In the last case, the mother was found to be an SMA carrier, and her husband was found to have two copies of SMN1 exon 7. However, the husband's father was an SMA deletion carrier, and his mother had a cis configuration (2/1 genotype). This meant that the husband had a 50% risk of being an SMA carrier, and the fetus had a 12.5% chance of being affected. Fortunately, chorionic villus sampling demonstrated that the fetus was unaffected.
In addition, during the study period, 11 more SMA carrier couples, previously ascertained to have a family history of SMA, underwent prenatal diagnosis (10 by chorionic villus sampling and 1 by amniocentesis). One fetus was found to be affected and pregnancy was terminated. Finally, during this period, one SMA carrier couple underwent successful preimplantation genetic diagnosis for SMA, resulting in the birth of healthy offspring.
SMA population-based screening was introduced in Israel in 2007. At that time, more than 15 other screening tests were already being offered.23 Some of these tests (cystic fibrosis [CF], Tay Sachs, and familial dysautonomia) were provided free of charge by the Ministry of Health. In this setting, we sought to evaluate the uptake rate of the newly added SMA screening test. We considered only patients undergoing genetic screening for the first time. We included only individuals who had requested at least two other screening tests that were recommended by the Israeli Society of Medical Genetics for people of all ethnic backgrounds (CF and fragile X syndrome). Both of these tests have been offered in Israel for more than 15 years. They are the mainstay of prenatal genetic screening and are highly accepted by the Israeli population. As previously noted, CF screening is provided at no cost, whereas fragile X testing is partially subsidized, with a copay between $25 and $100. The cost of SMA testing during the study period was also between $25 and $100. Among the 3606 women who underwent both CF and fragile X testing between September 2007 and August 2009, a total of 3336 (93%) requested SMA testing as well. Interestingly, during the first 4 months of the study, only 76% of the women requested SMA testing, while toward the end of the study period, this number increased to 99%. These data suggest that the Israeli population has accepted the newly introduced SMA screening to the same extent as they accept other recommended genetic screening tests.
DISCUSSION
Population-based screenings programs are offered worldwide for a number of common genetic disorders, such as cystic fibrosis and Tay Sachs disease. Several criteria have been proposed for choosing genetic disorders appropriate for population-based screening. These criteria include severity, early onset, high frequency of carriers in the screened population, availability of genetic counseling and prenatal diagnosis, and an existence of a reliable test.24
As a relatively common lethal autosomal recessive disorder with a high carrier frequency (1:40 in our population), SMA is a good candidate for population-based screening. Indeed, SMA screening was added to the recommended population screening tests in Israel in 2007; and in 2008, SMA carrier testing was recommended by the American College of Medical Genetics for all couples regardless of race or ethnicity.7 Here, we report our experience in initiating a large-scale population-based screening program for SMA.
The SMA screening tests that are currently used are based on dosage analysis; thus, those carriers that carry two or more SMN1 copies on the same chromosome (cis configuration) will not be detected. Such individuals still have a 50% chance of transferring the deleted allele to their offspring. Given the fact that cis configuration occurs in about 11% of individuals tested in our series, we estimate that about 1:760 reported as not being an SMA carrier by virtue of having 2 SMN1 copies shall in fact be SMA carriers. In other words, about 5.5% of SMA carriers shall have 2/0 genotype and will not be detected. This rate is higher than previously estimated. Feldkotter et al.,3 using real-time polymerase chain reaction, estimated that cis configuration occurs in only 4.8%, and Ogino et al.25 found cis configuration in about 7% of tested individuals. Recent studies of larger series performed in ethnically different populations estimated that the 2/0 genotype may account for as many as 8–8.8% of SMA carriers.18,19 These differences may be a result of different methods used to detect the copy number or be due to ethnic variations.
It has been estimated that about 6% of patients with SMA have either a de novo deletion or an inherited intragenic mutation.5,6,16 Given the fact that these too will not be detected by any quantitative screening methods, the detection rate for SMA carriers is unlikely to exceed 90%.
From a practical point of view, individuals in our population found to be SMA carriers who have a partner with two SMN1 copies still carry a residual risk of about 1:3000 for SMA affected offspring. This is obviously a small, but not negligible risk (especially, when one considers that if both partners have normal results, the residual risk is <1:100,000). In such circumstances, it may be possible to further reduce the remaining risk by testing both parents of the apparently normal spouse. If this individual is indeed a 2/0 SMA carrier, then usually one of his or her parents will carry a deletion (1/0 genotype) while the other will manifest three or more SMN copies (2/1 genotype). If this scenario is encountered, the “apparently normal” partner has a 50% chance of being a carrier, and prenatal diagnosis is indicated. Such a strategy will increase the carrier detection efficiency to about 95%, albeit at a significantly greater cost. This strategy has been used in a single case, as reported in the Results section.
Alternatively, when one partner is diagnosed as a carrier and the other has a normal result, prenatal diagnosis may be considered, as it will detect affected fetuses of parents with 2/0 genotype and also those affected by a de novo deletion. Such approach has been adopted by two couples, where one was detected as an SMA carrier and the other was screen-negative. This approach will also increase the detection rate to about 95% but at the price of procedure-related pregnancy losses. However, this may be considered when invasive prenatal testing is performed for another indication, such as advanced maternal age.
Although our study does not deal with the cost effectiveness of SMA prenatal screening, its results may impact on further such studies. The SMA carrier rate in our study (1:40) suggests a disease prevalence of 1:6400. This implies that the cost of averting a single case will be 36% lower than that based on a prevalence of 1:10,000, as estimated by Little et al.26 who claim that SMA prenatal screening is not cost effective. Moreover, the cost of SMA screening in Israel is currently much lower than that in the United States, partially because of the fact that SMA screening in Israel is usually performed as a part of a comprehensive screening panel and therefore has much lower marginal costs. These differences may impact upon cost effectiveness of SMA screening.
Finally, this study underscores the importance of defining clinically applicable cutoff values for quantitative analyses, such as MLPA. As no clear demarcation exists between RPA ratios corresponding to 1 vs. 2 SMN1 exon 7 copies, it may be prudent to use the conservatively higher cutoff of 0.75 to increase sensitivity, while only minimally increasing false positives.
In summary, our experience from this large-scale population-based program confirms that SMA screening is feasible and well acceptable among Israeli population. Moreover, we have shown that the newly introduced SMA screening test was quickly accepted by the Israeli population to the same extent as other recommended screening tests. Despite some inherent limitations, SMA screening is expected to detect the majority of at-risk couples. This may provide empowering reproductive options to couples, such as prenatal diagnosis and preimplantation genetic diagnosis.27
REFERENCES
OMIM (Online Mendelian inheritance in man) Baltimore: Johns Hopkins University, Center for Medical Genetics, 1996. Available at: http://www.ncbi.nlm.nih.gov/omim.
Pearn J . Classification of spinal muscular atrophies. Lancet 1980; 1: 919–922.
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Ogino S, Wilson RB, Gold B . New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet 2004; 12: 1015–1023.
Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165.
Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999; 64: 1340–1356.
Prior TW ; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med 2008; 10: 840–842.
American College of Obstetricians and Gynecologist. ACOG committee opinion, No. 432: spinal muscular atrophy. Obstet Gynecol 2009; 113: 1194–1196.
Burglen L, Lefebvre S, Clermont O, et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996; 32: 479–482.
Lorson CL, Hahnen E, Androphy EJ, Wirth B . A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 1999; 96: 6307–6311.
McAndrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997; 60: 1411–1422.
Scheffer H, Cobben JM, Mensink RG, Stulp RP, van der Steege G, Buys CH . SMA carrier testing—validation of hemizygous SMN exon 7 deletion test for the identification of proximal spinal muscular atrophy carriers and patients with a single allele deletion. Eur J Hum Genet 2000; 8: 79–86.
Tomaszewicz K, Kang P, Wu BL . Detection of homozygous and heterozygous SMN deletions of spinal muscular atrophy in a single assay with multiplex ligation-dependent probe amplification. Beijing Da Xue Xue Bao 2005; 37: 55–57.
Arkblad E, Darin A, Berg K, et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord 2006; 12: 830–838.
Scarciolla O, Stuppia L, De Angelis MV, et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006; 7: 269–276.
Chen KL, Wang YL, Rennert H, et al. Duplications and de novo deletions of the SMNt gene demonstrated by fluorescence-based carrier testing for spinal muscular atrophy. Am J Med Genet 1999; 85: 463–469.
Wirth B, Schmidt T, Hahnen E, et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61: 1102–1111.
Smith M, Calabro V, Chong B, Gardiner N, Cowie S, du Sart D . Population screening and cascade testing for carriers of SMA. Eur J Hum Genet 2007; 7: 759–766.
Zhu S, Xiong F, Chen YJ, et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet 2010; 18: 978–984.
Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A 2010; 152: 1608–1616.
Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 2009; 46: 641–644.
Sukenik-Halevy R, Pesso R, Garbian N, Magal N, Shohat M . Large-scale population carrier screening for spinal muscular atrophy in Israel—effect of ethnicity on the false-negative rate. Genet Test Mol Biomarkers 2010; 14: 319–324.
Rosner G, Rosner S, Orr-Urtreger A . Genetic testing in Israel: an overview. Annu Rev Genomics Hum Genet 2009; 10: 175–192.
Statement of American Society of Human Genetics on cystic fibrosis carrier screening. Am J Hum Genet 1992; 51: 1443–1444.
Ogino S, Leonard DG, Rennert H, Ewens WJ, Wilson RB . Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet 2002; 110: 301–307.
Little SE, Janakiraman V, Kaimal A, Musci T, Ecker J, Caughey AB . The cost-effectiveness of prenatal screening for spinal muscular atrophy. Am J Obstet Gynecol 2010; 202: 253.e1–253.e7.
Malcov M, Schwartz T, Mei-Raz N, et al. Multiplex nested PCR for preimplantation genetic diagnosis of spinal muscular atrophy. Fetal Diagn Ther 2004; 19: 199–206.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ben-Shachar, S., Orr-Urtreger, A., Bardugo, E. et al. Large-scale population screening for spinal muscular atrophy: Clinical implications. Genet Med 13, 110–114 (2011). https://doi.org/10.1097/GIM.0b013e3182017c05
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3182017c05
Keywords
This article is cited by
-
Population WGS-based spinal muscular atrophy carrier screening in a cohort of 1076 healthy Polish individuals
Journal of Applied Genetics (2023)
-
Health Care Resource Utilization and Costs for Patients with Spinal Muscular Atrophy: Findings from a Retrospective US Claims Database Analysis
Advances in Therapy (2023)
-
Accurate diagnosis of spinal muscular atrophy and 22q11.2 deletion syndrome using limited deoxynucleotide triphosphates and high-resolution melting
BMC Genomics (2018)
-
Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review
Orphanet Journal of Rare Diseases (2017)
-
Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients
Journal of Human Genetics (2015)
