
Abstract
Genomic microarrays used to assess DNA copy number are now recommended as first-tier tests for the postnatal evaluation of individuals with intellectual disability, autism spectrum disorders, and/or multiple congenital anomalies. Application of this technology has resulted in the discovery of widespread copy number variation in the human genome, both polymorphic variation in healthy individuals and novel pathogenic copy number imbalances. To assist clinical laboratories in the evaluation of copy number variants and to promote consistency in interpretation and reporting of genomic microarray results, the American College of Medical Genetics has developed the following professional guidelines for the interpretation and reporting of copy number variation. These guidelines apply primarily to evaluation of constitutional copy number variants detected in the postnatal setting.
Similar content being viewed by others

Main
Genomic microarrays used to assess DNA copy number, often referred to as chromosomal or cytogenetic/cytogenomic microarrays, are powerful clinical diagnostic tools now recommended as first-tier tests for the postnatal evaluation of individuals with intellectual disability, autism spectrum disorders, and/or multiple congenital anomalies.1,2 Most genomic microarrays used in clinical practice, including array-based comparative genomic hybridization and single-nucleotide polymorphism-based platforms, provide genome-wide coverage for detection of chromosomal imbalances at a much higher resolution than a conventional cytogenetic analysis (e.g., G-banded karyotype). The ability to examine the genome at this high resolution has resulted in the discovery of widespread copy number variation in the human genome, both polymorphic variation in healthy individuals and novel pathogenic copy number imbalances.
A copy number variant (CNV) is defined as a segment of DNA at least 1 kb in size that differs in copy number compared with a representative reference genome. The term “CNV” does not imply clinical significance; therefore, a qualifier such as pathogenic CNV or benign CNV is necessary for clear communication of clinical relevance. In addition, the term “CNV” does not imply relative dosage. Copy number loss (deletion) or copy number gain (duplication) must be specified to clarify the nature of a CNV.
Although genomic regions rich in low copy repeat sequences (or segmental duplications) may result in commonly recurring CNVs usually well described in the medical literature, many CNVs represent rare variation. In many cases, the interpreting geneticist can assess the genomic content of the CNV, correlate with established clinical literature, and provide an interpretation that is unambiguous and consistent with the interpretation derived from multiple laboratories.3 However, given the presence of benign CNVs within the genome and the continual discovery of novel CNVs, assessing the clinical significance of CNVs found in a clinical setting can be challenging.4,5 Accordingly, when the CNV is extremely rare or limited clinical literature is available, interpretation and reporting practices may vary among laboratories.6 To assist clinical laboratories in the evaluation of CNVs and to promote consistency in interpretation and reporting of genomic microarray results, the American College of Medical Genetics (ACMG) has developed the following professional guidelines for the interpretation and reporting of copy number variation. In addition, these guidelines may serve as a reference for referring clinicians, so they may better understand the complexity of CNV interpretation and communicate these findings to patients and families appropriately.
These guidelines apply primarily to evaluation of constitutional CNVs detected in the postnatal setting. Although these guidelines are relevant for CNVs detected during prenatal testing, interpretation and reporting of prenatal CNVs require additional considerations outside the scope of this document. These guidelines do not apply to CNVs representing acquired mutations in neoplasia. In addition, this document does not address analytical validation; therefore, the recommendations assume that the laboratory is confident the CNV represents true biological copy number variation in the patient. Although these guidelines attempt to cover common issues encountered during evaluation of CNVs, there are many CNVs with unique characteristics, and no algorithm will be applicable to all findings.
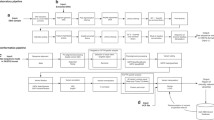
RECOMMENDATIONS FOR SYSTEMATIC EVALUATION AND CLINICAL INTERPRETATION OF CNVs
Familiarization with well-established contiguous gene syndromes
Approaching a large segmental deletion or duplication by interrogation of single genes within the interval may not reveal the associated syndromic deletion/duplication. It is necessary that recurrent and clinically characterized deletion/duplication syndromes, any associated low copy repeat sequences, and critical regions be recognized and carefully mapped before offering clinical interpretations of microarray data. The following represent helpful reviews but do not substitute for continuing education and monitoring of the rapidly expanding medical literature: OMIM,7 GeneReviews,8 DECIPHER,9 and recently published reviews.10,11
Consideration of CNV size
Although generalizations drawn between CNV size and significance hold true as a general rule, it is clear that very large CNVs can be benign in nature,12–15 and very small CNVs can be clinically significant. It is, therefore, recommended that any size restriction for inclusion of a CNV in a clinical report be based on the laboratory's consideration of the performance characteristics of the array used and generation of a reasonable amount of clinical follow-up, rather than assumptions regarding clinical significance.
Consideration of genomic content in CNV interval
This is by far the most relevant interpretive consideration and is fairly broad in scope. One should consider whether the CNV contains unique, gene-rich sequence or is void of genes and/or is primarily comprised of repetitive elements or pseudogenes. The gene content should be carefully scrutinized for documented and relevant clinical association.
When considering the potential phenotypic effect due to copy number gain or loss of a gene or group of genes, one should investigate whether the genes in the interval are reported to be dosage sensitive and associated with clinical disorders. It is useful and convenient to review entries in the OMIM database7; however, it is also prudent to search for recent publications that may not have been incorporated into the OMIM review.
Given that CNV breakpoints are not precisely mapped due to gaps in probe coverage, it is important to consider all the genes in the maximum CNV interval before presuming a CNV to be clinically benign. Further evaluation may be necessary to clarify the genomic content of the CNV for appropriate clinical interpretation.
Genes with reported pathogenic mutations in the medical literature.
The nature of the disease-associated mutations should be carefully interrogated to ensure relevance for the CNV in question. Although this list is not comprehensive, the following examples illustrate the need for a clear understanding of genetic mechanisms: (1) a gene associated with a clinical phenotype due to haploinsufficiency may have no phenotype associated with a copy number gain. (2) Dominant disorders often result from specific gain of function mutations rather than dosage imbalance; therefore, CNVs involving such genes may either have no clinical relevance or result in an entirely different phenotype (e.g., gain of function/activating mutations in FGFR1 result in skeletal dysplasias, whereas deletions/loss of function mutations are associated with Kallmann syndrome16). (3) Copy number gains involving only part of a gene may result in gene disruption or altered coding sequence and should not be dismissed without further investigation when involving genes with reported haploinsufficiency.17 (4) Single-copy deletions of genes associated with recessive disease may only suggest carrier status for the condition. (5) Small CNVs involving only intronic sequence may have no effect on gene function.
Genes with no reported mutations in the medical literature.
Avoid, or use great caution when, inferring a pathogenic role for a gene based solely on predicted gene function or functions characterized in model organisms or in vitro studies. This inference is speculative until well characterized in the human population.
No genes in interval.
Generally, it is acceptable to adopt a laboratory policy not to report these CNVs, as there is no relevant literature to interrogate. An exception might be made if the CNV exceeds a size cutoff established by the laboratory or is located in close proximity to a well-characterized region with clear relevance to the reason for referral (e.g., a deletion bordering a holoprosencephaly locus in a patient with a holoprosencephaly indication18–20).
Comparison of CNV with internal and external databases
We recommend that laboratories performing array-based assessment of copy number track their experience and document pathogenic CNVs, CNVs of uncertain significance, and CNVs that have been determined to represent benign variation. A CNV that is well documented as a benign variant in the performing laboratory, or in peer reviewed published reports or curated databases, likely needs little additional investigation provided the CNV is periodically reinvestigated to ensure that no new data have emerged contradicting this classification. A CNV with which the laboratory has no prior experience should be carefully compared with publicly available databases of copy number variation in the general population, such as the Database of Genomic Variants.21,22 Note that we have used the term general population rather than normal population, as it is clear that “normal” is relative to the phenotype in question, and some of the individuals represented in large population databases have no phenotypic data available (e.g., HapMap).
Considerations when comparing patient data with CNVs reported in general population databases include:
Dosage of copy number imbalance reported in the general population studies.
The CNV reported as a benign finding in the general population might exclusively represent a copy number gain. If the CNV in question overlaps the same region, but is a copy number loss, a pathogenic outcome cannot be excluded. The opposite situation also applies. Similarly, a CNV observed commonly as a heterozygous deletion in the general population may have a pathogenic outcome when present in a homozygous state.
Size of the reported benign CNV relative to the CNV in question.
One should ensure that the CNV reported in the general population includes the same gene content as the CNV being interrogated. Different array platforms represented in the public databases can lead to differences in the reported size of identical CNVs. Notably, many of the benign CNVs reported from earlier bacterial artificial chromosome-based microarray studies represent size overestimates.23
Sex of individual in database relative to patient sex.
This consideration is particularly important for X-linked CNVs in males, as many of the reported benign variants are seen in females who may be nonmanifesting carriers of the condition. In addition, consider that contributors to these databases may have excluded the sex chromosomes from analysis; therefore, CNVs mapping to the X and Y chromosomes may be underrepresented.
Validity of the CNV reported in general population databases.
The majority of CNVs reported from large population studies have not been experimentally validated; therefore, CNVs reported in a single study or through use of a single microarray platform should be interpreted with caution.
Clinical characterization of “normal” individuals.
One should consider the extent of clinical characterization of individuals represented in the database. Each population series is selected based on defined criteria, usually outlined in the primary reference. Consider how these individuals were selected for inclusion and how likely it is that the clinical phenotype presented in the patient of interest might be present in the “normal” population. Factors such as incomplete penetrance, variable expressivity, age of onset, and parent of origin imprinting effects need to be considered before classification of a CNV as benign in all instances. CNVs occurring with relatively high frequency in the general population and in multiple publications may be interpreted with more confidence as benign in nature. Of note, many publications use the same reference set (e.g., HapMap); therefore, a CNV represented in multiple publications may represent the same individual studied multiple times.
RECOMMENDED CATEGORIES OF CLINICAL SIGNIFICANCE
Using the guidelines outlined earlier for systematic investigation of a CNV for clinical significance, the interpreting laboratory geneticist should assign any CNV reported in the patient to one of three main categories of significance. It is recommended that consistent terminology for these categories be used in clinical reporting to facilitate unambiguous communication of clinical significance throughout the medical community.
Pathogenic
The CNV is documented as clinically significant in multiple peer-reviewed publications, even if penetrance and expressivity of the CNV are known to be variable.
This category includes large CNVs, which may not be described in the medical literature at the size observed in the patient but which overlap a smaller interval with clearly established clinical significance. Although the full clinical effect of the patient's CNV is not known, the pathogenic nature of the CNV is not in question.
With the exception of well-established cytogenetic heteromorphisms, this category will include most cytogenetically visible alterations (>3–5 Mb). In the absence of clearly defined syndromic loci within the interval, this inference should be made with caution.
Uncertain clinical significance
This represents a fairly broad category and will include findings that are later demonstrated to be either clearly pathogenic or clearly benign. However, if at the time of reporting, insufficient evidence is available for unequivocal determination of clinical significance and the CNV meets the reporting criteria established by the laboratory, the CNV should be reported as a CNV of uncertain clinical significance.
When warranted, one may provide evidence for the likelihood that the CNV is pathogenic or benign, provided any such speculation is well supported in the report, and the uncertainty of this classification is still communicated. Three categories for classification of uncertain variants are suggested and examples provided below. These examples do not cover all scenarios, as each CNV will have unique considerations requiring clinical judgment.
-
Uncertain clinical significance; likely pathogenic: for example: (1) The CNV is described in a single case report but with well-defined breakpoints and phenotype, both specific and relevant to the patient findings. (2) A gene within the CNV interval has a very compelling gene function that is relevant and specific to the reason for patient referral. Inferences made from data derived solely from model systems should be made with discretion and in general, are discouraged. Such speculation is strongly discouraged for scenarios with nonspecific indications (e.g., intellectual disability) and/or limited evidence regarding gene function (e.g., only information available for gene is documented neuronal expression).
-
Uncertain clinical significance; likely benign: for example: (1) The CNV has no genes in interval (but is reported because it exceeds a size criterion that may be established by the laboratory). (2) The CNV is described in a small number of cases in databases of variation in the general population but does not represent a common polymorphism.
-
Uncertain clinical significance (no subclassification): for example: (1) The CNV contains genes, but it is not known whether the genes in the interval are dosage sensitive. (2) The CNV is described in multiple contradictory publications and/or databases, and firm conclusions regarding clinical significance are not yet established.
Benign
The CNV has been reported in multiple peer-reviewed publications or curated databases as a benign variant, particularly if the nature of the copy number variation has been well characterized (e.g., copy number variation of the salivary amylase gene24) and/or the CNV represents a common polymorphism. To qualify as a polymorphism, the CNV should be documented in >1% of the population. It is important to carefully consider dosage of the CNV documented as a benign variant, given, for example, that duplications of some regions may be benign, whereas deletions of the same interval may have clinical relevance.
REPORTING GUIDELINES FOR GENOMIC COPY NUMBER MICROARRAYS USED IN THE POSTNATAL CONSTITUTIONAL SETTING
The following guidelines describe those elements of the clinical report that are necessary to specify the precise identity of a CNV and clearly communicate the clinical significance of the microarray results. Other required elements of a clinical report (e.g., methodology and relevant disclaimers) are outlined in detail in the ACMG Laboratory Standards and Guidelines.
Reporting criteria
The laboratory report should include a description of the criteria used to review the data (e.g., CNV size restrictions) and the criteria used for inclusion of a CNV in the report. Laboratories may choose not to disclose benign CNVs, especially those that represent common polymorphisms. If benign CNVs are listed in the report, they should meet the same analytical performance criteria used in the laboratory to evaluate suspected pathogenic CNVs.
Description of position, size, and relative gain or loss for each CNV detected
Each CNV included in the laboratory report should be described with the elements below. Current nomenclature from the International System for Cytogenetic Nomenclature should be included in the report but should not serve as a substitute for a clear description of the imbalance for clinical professionals unfamiliar with International System for Cytogenetic Nomenclature conventions.
-
Cytogenetic location (chromosome number and cytogenetic band designation).
-
Dosage (e.g., copy number gain or loss) with CNV mechanism specified when understood (e.g., single-copy deletion, tandem duplication). Assessment of mechanism will usually require additional testing methodology.
-
CNV size and linear coordinates with genome build specified. When applicable, particularly when gene content of the CNV is unclear, minimum/maximum coordinates should be provided.
Clear statement of clinical significance
Each reported CNV should be accompanied by a clear statement of significance as outlined in these recommendations. The evidence in support of this interpretation should be summarized and appropriate references provided.
Designation of genes in CNV interval
To the extent feasible, genes involved in a CNV should be specified in the laboratory report. For large imbalances, particularly those with well-established clinical significance, it is acceptable to provide only the name of the corresponding syndrome and/or the most clinically relevant genes in the interval. For CNVs of uncertain significance, it is suggested that all RefSeq genes in the interval be included in the report, when feasible, to facilitate periodic reviews of relevant medical literature. The incorporation of links to websites that list the genes in a particular interval is not recommended because the links may not faithfully direct the clinician to the appropriate gene content in the future, particularly when genome builds change. When only one or few representative genes are specified in the report, it is suggested that the total number of genes in the CNV interval be provided to allow for perspective of the total gene content.
Recommendation for appropriate clinical follow-up
When pathogenic CNVs or CNVs of uncertain clinical significance are found, the laboratory report should include recommendations for genetic consultation/counseling, any necessary cytogenetic characterization of the CNV, and evaluation of relevant family members. In addition, when a CNV is determined to have uncertain clinical significance, the report may include a recommendation for continued surveillance of the medical literature for new information, which could later clarify the clinical significance of the finding. The responsibility for continuing monitoring of the medical literature for a specific patient lies primarily with the physician with an ongoing patient relationship.25
SPECIAL CONSIDERATIONS REGARDING REPORTING OF UNANTICIPATED CLINICALLY SIGNIFICANT FINDINGS UNRELATED TO THE REASON FOR REFERRAL
In rare cases, CNVs may be found in a patient which (1) reveal carrier status for a recessive condition, (2) are diagnostic or predictive of a presymptomatic condition or a symptomatic condition with unrecognized clinical presentation, or (3) are associated with an increased risk of neoplasia. In general, these findings are unanticipated and unrelated to the patient's reason for referral for a genomic screen. It is not possible to construct a whole genome microarray platform that purposefully avoids interrogation of any loci associated with the aforementioned cases, especially as many of the findings will be part of a large CNV involving multiple contiguous genes. Therefore, referring clinicians must have a clear understanding of the potential for these discoveries, and patients/families should be duly informed before test ordering. A formal informed consent process is strongly recommended. In rare situations, creation of unique CNV-reporting categories (e.g., carrier-presumed unaffected) may be necessary for unambiguous reporting.
Reporting carrier status for recessive conditions
It is recognized that detection of some CNVs, particularly deletions, will reveal carrier status for recessive disorders in the deletion interval. Comprehensive reporting of heterozygous recessive mutations is outside the scope of the intended use of these tests and, in general, is not recommended. Reports should clearly state that recessive carrier status may not be disclosed, and that any clinical concern for recessive disorders should be communicated to the reporting laboratory for appropriate consideration. Individual laboratories may choose to adopt specific disclosure policies for recessive conditions. There are some situations when disclosure of recessive mutations may be considered.
-
1
Well-characterized recessive disorders, for which carrier frequency is reasonably high in the patient population and/or carrier screening is commonly available (e.g., cystic fibrosis). In such cases, there may be justification for reporting carrier status to provide opportunity for reproductive counseling and potential additional testing in the proband or relevant family members. It should be recognized that these disclosures will represent serendipitous findings, and no claim should be made to the ordering clinician or patient that this test will detect carrier status for any condition.
-
2
Recessive disorders with clinical features consistent with the patient's reason for referral. In such cases, recommendation for further molecular testing for this disorder may be warranted. This should be restricted to well-described recessive disorders with clear clinical consequence. The report should clearly state the recessive nature of the condition and that the CNV is not diagnostic of affected status without confirmation of a second mutation.
Reporting mutation status for adult onset/ presymptomatic or undiagnosed conditions
Some CNVs, although unrelated to the patient's reason for referral, may be clearly diagnostic of a presymptomatic or clinically undetected condition (e.g., male infertility due to deletions involving the AZF region on the Y chromosome26). Given that it is impossible to construct a predefined list of all possible diagnoses to allow the patient to consent specifically to the interrogation/reporting of each disorder, it is our general recommendation that CNVs associated with presymptomatic conditions be reported to facilitate early access to medical care. Individual laboratories may wish to adopt nondisclosure policies for specific conditions. Any such policies should be stated in the laboratory report.
Reporting CNVs associated with risk of neoplasia
Deletions that include a known or putative tumor suppressor gene should be carefully considered. Tumor suppressor genes with clearly pathogenic germline mutations and information on penetrance, lifetime risk, tumor spectrum, and clinical management (e.g., RB1, TP53, and APC) should be discussed in the report, regardless of the indication for study.27 Speculation regarding putative tumor suppressor genes should be avoided, particularly in the absence of well-characterized germline mutations in humans and in the case of genes where tumor suppressor functions have only been shown in animal or in vitro models.
SPECIAL CONSIDERATIONS FOR REEVALUATION OF SIGNIFICANCE BASED ON DATE FROM OTHER FAMILY MEMBERS
When CNVs are found to have uncertain clinical significance, it may be informative to determine whether the CNV was inherited or represents a de novo mutation. Although this is a very relevant line of evidence by which to gauge clinical significance, it is important to stress that it is difficult, and often imprudent, to attribute clinical significance based on the inheritance pattern of a CNV in a single family. It is only through ascertainment of significantly large families with multiple affected and unaffected family members segregating a given CNV or ascertainment of multiple individuals with the same CNV that a true measure of clinical significance can be confidently assessed. For this reason, the ACMG strongly supports efforts to collect and curate deidentified patient data from clinical studies to facilitate rapid and unambiguous assessment of the clinical significance of CNVs.2,9,28
In the absence of a large data series, a cautious inference should be made based on the limited family information available. Addendums to the original interpretation, based on information from evaluation of family members, should be appropriately communicated with disclaimers summarizing the following points. Each CNV and each family will have unique considerations, and these investigations require significant communication between laboratorians and referring clinicians.
De novo CNVs
When a CNV is shown to represent a de novo mutation in the proband, this is generally taken as evidence supporting pathogenicity, particularly if the CNV was suspected to have clinical significance based on other lines of evidence, such as gene content. Nonpaternity may complicate this interpretation and, as such, should be disclaimed in the report. Specific testing to confirm paternity is not recommended unless there is a compelling clinical reason to make this assessment and explicit informed consent is obtained.
Because many regions of the genome have significantly elevated mutation rates, some CNVs may indeed be de novo mutations yet represent findings with no associated clinical consequence.29
If only one parent is available for follow-up, and the CNV is not found in the parent available for study, no additional inference regarding clinical significance can be made.
Inherited CNVs
When a CNV is found in a parent or other relevant family member, there are numerous caveats one should consider. Rarely can a conclusive inference be made based on the inheritance pattern observed in a single family. The carrier parent and other relevant family members should have a thorough medical evaluation for the presence or absence of the clinical features present in the proband. When this information is not provided to the laboratory, this should be disclaimed in the report and a recommendation for correlation with parental clinical features made.
Parent is affected.
In general, this may be cautiously taken as evidence that supports the CNV as the cause of the clinical features. This observation may be coincidental, however, as the CNV and clinical trait may be inherited independently by chance. When available, other family members may be evaluated to determine whether the CNV continues to segregate in concordance with the clinical phenotype.
Parent is unaffected.
In general, this may be taken as evidence that supports the CNV as unrelated to the clinical features and likely benign. Special considerations that preclude confident inference and may only be well defined after ascertainment of multiple families include the following:
-
Incomplete penetrance: The CNV may be pathogenic but nonpenetrant in the carrier parent.
-
Variable expressivity: The carrier parent may have subclinical features that will later be shown to be in the spectrum of the disorder caused by the CNV.
-
Parent of origin imprinting effects: The CNV region may be imprinted, such that the disorder only manifests when inherited from a particular sex (and the carrier parent is not manifesting the disorder because of chance inheritance).
-
Second mutation not detectable by microarray: The proband may be manifesting a recessive disorder (e.g., a deletion may be inherited from an unaffected carrier parent and an undetectable mutation inherited in a gene within the interval from the other parent). Alternatively, the proband may have one or more “modifier” genes/DNA elements not present in the unaffected parent.
-
Mosaic CNV in parent: The CNV may not be present in all tissues of the parent, and therefore, the parent may not manifest all clinical features associated with the CNV.
-
CNV in proband is not identical in size to that seen in parent: Rarely, CNVs have been found to undergo further modification (e.g., expansion of deletion) when transmitted from carrier parent to affected child.30 When the parental studies are performed by an alternative methodology such as fluorescence in situ hybridization, this rare possibility cannot be excluded.
-
Special consideration for X-linked CNVs: When an X-linked CNV in a male is found in an unaffected carrier mother, one should consider whether the mother is a nonmanifesting carrier. Studies of X-inactivation may be informative, but not all X-linked disorders exhibit skewed X-inactivation in carrier females. It may be more informative to seek other male relatives in the maternal family.
CONCLUDING REMARKS
With the expanding availability of whole-genome testing methodologies, clinical genetics professionals must be prepared to interpret unexpected findings and report them appropriately, not only with consideration of clinical relevance but also with consideration of social, ethical, and legal responsibilities. The interpretation of the clinical relevance of copy number variation is complex and is the practice of medicine. As evident from the numerous considerations outlined in this document, there is no one formula or algorithm for CNV interpretation that will substitute for adequate training in clinical genetics and sound clinical judgment. We, therefore, recommend that the reporting of clinical genomic microarrays be performed by individuals with appropriate professional training and certification (American Board of Medical Genetics-certified clinical cytogeneticists, American Board of Medical Genetics-certified clinical molecular geneticists, or American Board of Medical Genetics/American Board of Pathology-certified molecular genetic pathologists). In addition, given the complexity of this interpretation and the different laboratory methodologies necessary for CNV characterization and evaluation of additional family members, the ideal laboratory setting for this testing is one with both cytogenetic and molecular genetic expertise.
References
Manning M, Hudgins L, Professional Practice and Guidelines Committee Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med 2010; 12: 742–745.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010; 86: 749–764.
Brothman AR, Dolan MM, Goodman BK, Park JP, Persons DL, Saxe DF, et al. College of American Pathologists/American College of Medical Genetics proficiency testing for constitutional cytogenomic microarray analysis. [published online ahead of print May 30, 2011]. Genet Med doi: 10.1097/GIM.0b013e31821d3165
Lee C, Iafrate AJ, Brothman AR . Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet 2007; 39( suppl 7): S48–S54.
Sharp AJ . Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat 2009; 30: 135–144.
Tsuchiya KD, Shaffer LG, Aradhya S, et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med 2009; 11: 866–873.
McKusick VA National Center for Biotechnology Information (U.S.). OMIM. Bethesda, MD: The Center, 1987. Available at: http://www.ncbi.nlm.nih.gov/Omim/. Accessed April 24, 2011.
Pagon RA GeneReviews. Seattle: University of Washington. Available at: http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call_bv.View.ShowTOC&rid_gene.TOC. Accessed April 24, 2011.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009; 84: 524–533.
Slavotinek AM . Novel microdeletion syndromes detected by chromosome microarrays. Hum Genet 2008; 124: 1–17.
Mefford HC, Eichler EE . Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev 2009; 19: 196–204.
Bateman MS, Mehta SG, Willatt L, et al. A de novo 4q34 interstitial deletion of at least 9.3 Mb with no discernible phenotypic effect. Am J Med Genet A 2010; 152A: 1764–1769.
Itsara A, Cooper GM, Baker C, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 2009; 84: 148–161.
Filges I, Röthlisberger B, Noppen C, et al. Familial 14.5 Mb interstitial deletion 13q21.1–13q21.33: clinical and array-CGH study of a benign phenotype in a three-generation family. Am J Med Genet A 2009; 149A: 237–241.
Barber JC . Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet 2005; 42: 609–629.
Wilkie AO . Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev 2005; 16: 187–203.
Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS . Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet 2009; 46: 68–72.
Cooper DN, Chen JM, Ball EV, et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat 2010; 31: 631–655.
Kleinjan D, van Heyningen V . Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 2005; 76: 8–32.
Beysen D, Raes J, Leroy BP, et al. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet 2005; 77: 205–218.
Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Shaikh TH, Gai X, Perin JC, et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 2009; 19: 1682–1690.
Perry GH, Ben-Dor A, Tsalenko A, et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 2008; 82: 685–695.
Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet 2007; 39: 1256–1260.
Hirschhorn K, Fleisher LD, Godmilow L, et al. Duty to re-contact. Genet Med 1999; 1: 171–172.
Vogt PH . Genomic heterogeneity and instability of the AZF locus on the human Y chromosome. Mol Cell Endocrinol 2004; 224: 1–9.
Adams SA, Coppinger J, Saitta SC, et al. Impact of genotype-first diagnosis: the detection of microdeletion and microduplication syndromes with cancer predisposition by aCGH. Genet Med 2009; 11: 314–322.
Church DM, Lappalainen I, Sneddon TP, et al. Public data archives for genomic structural variation. Nat Genet 2010; 42: 813–814.
Bradley WE, Raelson JV, Dubois DY, et al. Hotspots of large rare deletions in the human genome. PLoS One 2010; 5: e9401.
South ST, Rope AF, Lamb AN, et al. Expansion in size of a terminal deletion: a paradigm shift for parental follow-up studies. J Med Genet 2008; 45: 391–395.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The authors declare no conflict of interest.
Disclaimer: These ACMG Standards and Guidelines are developed primarily as an educational resource for clinical laboratory geneticists to help them provide quality clinical laboratory genetic services. Adherence to these standards and guidelines is voluntary and does not necessarily assure a successful medical outcome. These Standards and Guidelines should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinical laboratory geneticist should apply his or her own professional judgment to the specific circumstances presented by the individual patient or specimen. Clinical laboratory geneticists are encouraged to document in the patient's record the rationale for the use of a particular procedure or test, whether or not it is in conformance with these Standards and Guidelines. They also are advised to take notice of the date any particular standard or guidelines was adopted and to consider other relevant medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
Rights and permissions
About this article
Cite this article
Kearney, H., Thorland, E., Brown, K. et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13, 680–685 (2011). https://doi.org/10.1097/GIM.0b013e3182217a3a
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3182217a3a
Keywords
This article is cited by
-
Chromosomal microarray testing yield in 829 cases of microcephaly: a clinical characteristics-based analysis for prenatal and postnatal cases
Archives of Gynecology and Obstetrics (2024)
-
Partial trisomy 9p and partial monosomy 7p of an infant inherited from maternal balanced translocation: a case report
BMC Pediatrics (2023)
-
Clinical utility of periodic reinterpretation of CNVs of uncertain significance: an 8-year retrospective study
Genome Medicine (2023)
-
Clinical diagnosis of genetic disorders at both single-nucleotide and chromosomal levels based on BGISEQ-500 platform
Human Genome Variation (2023)
-
Exome sequencing in individuals with congenital anomalies of the kidney and urinary tract (CAKUT): a single-center experience
European Journal of Human Genetics (2023)
