
Abstract
Purpose
Unexpected fetal abnormalities occur in 2–5% of pregnancies. While traditional cytogenetic and microarray approaches achieve diagnosis in around 40% of cases, lack of diagnosis in others impedes parental counseling, informed decision making, and pregnancy management. Postnatally exome sequencing yields high diagnostic rates, but relies on careful phenotyping to interpret genotype results. Here we used a multidisciplinary approach to explore the utility of rapid fetal exome sequencing for prenatal diagnosis using skeletal dysplasias as an exemplar.
Methods
Parents in pregnancies undergoing invasive testing because of sonographic fetal abnormalities, where multidisciplinary review considered skeletal dysplasia a likely etiology, were consented for exome trio sequencing (both parents and fetus). Variant interpretation focused on a virtual panel of 240 genes known to cause skeletal dysplasias.
Results
Definitive molecular diagnosis was made in 13/16 (81%) cases. In some cases, fetal ultrasound findings alone were of sufficient severity for parents to opt for termination. In others, molecular diagnosis informed accurate prediction of outcome, improved parental counseling, and enabled parents to terminate or continue the pregnancy with certainty.
Conclusion
Trio sequencing with expert multidisciplinary review for case selection and data interpretation yields timely, high diagnostic rates in fetuses presenting with unexpected skeletal abnormalities. This improves parental counseling and pregnancy management.
Similar content being viewed by others

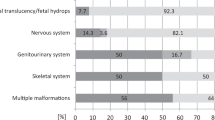
Introduction
Unexpected fetal abnormalities occur in around 2–5% of pregnancies1 and, while diagnosis can be achieved in up to 40% of cases using traditional cytogenetic and microarray approaches,2 the majority remain undiagnosed, many of which have a genetic etiology.3 Definitive genetic diagnosis of abnormalities during an ongoing pregnancy is challenging in the absence of a family history. It is generally only possible and timely in selected conditions where the phenotype is well described and there are limited numbers of disease-causing variants, for example, skeletal abnormalities caused by the FGFR2 and 3 pathogenic variants.4 Inability to make a definite diagnosis makes parental counseling and decision making difficult, complicates pregnancy management, and results in an emotional burden for couples.5
In the postnatal setting, exome sequencing is now part of routine clinical genetic practice,6 adding around 25–30% to the diagnostic yield in pediatric and adult patients suspected to have a genetic condition but with no diagnosis after traditional testing.7 However, limited data are available with regard to performance of exome sequencing for the diagnosis of fetal abnormalities although several small series of selected cases report diagnostic rates of between 6.2 and 80%.8 The majority of these studies report small numbers of selected cases with results returned to parents after the pregnancy ended. Nevertheless, the use of exome sequencing for the prenatal diagnosis of the dysmorphic fetus using trio testing of parents and abnormal fetus is gaining momentum,9,10 albeit with lower diagnostic yields of around 6–8% reported in larger consecutive case series of unselected fetuses.9,10
Prenatal investigations need to deliver accurate results with a high detection rate and in a timely fashion if they are to be clinically useful for informing parental decision making and pregnancy management. Skeletal dysplasias are a complex group of disorders that are particularly challenging to diagnose in the prenatal setting as they are individually rare, many of the ultrasound findings are not necessarily pathognomic for a specific condition, and most arise de novo. Furthermore, they are very heterogeneous with variable outcomes, some of which can be severe even in the presence of relatively minor prenatal findings. Thus, with the exception of achondroplasia and thanatophoric dysplasia, which have distinctive prenatal findings and limited numbers of causative variants,4 definitive diagnosis must often await the result of postnatal investigations or postmortem examination, the latter of which is not always accepted by parents. Furthermore, definitive molecular diagnosis will be a prerequisite for novel in utero treatment trials such as the Boost Brittle Bones Before Birth study (BOOSTB4),11 which will offer in utero mesochymal stem cell therapy for osteogenesis imperfecta (OI) types III and IV.12 A high-throughput multigene sequencing approach is the only way to achieve rapid diagnoses in this time-limited situation.13 Here, we explored the use of targeted exome sequencing for the rapid genetic diagnosis of fetuses with a suspected skeletal dysplasia as an exemplar of how exome sequencing can be used as an aid to prenatal diagnosis and pregnancy management.
Materials and methods
This study was approved under “New methods of detecting problems in pregnancy” Research Ethics Committee reference 01/0095.
Patients
Pregnant women who had had, or who were undergoing, an invasive procedure to exclude chromosomal abnormalities following ultrasound detection of fetal abnormalities suggestive of a skeletal dysplasia in UK fetal medicine units were identified prospectively. Inclusion criteria included short long bones with or without evidence of bowing or fracturing of bones, hypomineralization, hydrops, or thoracic hypoplasia. Ultrasound images were reviewed by the local fetal medicine teams and clinical geneticists with expertise in fetal dysmorphology. Where there was agreement that the findings were suspicious of an underlying skeletal dysplasia, written informed consent was obtained to take parental blood and use excess fetal amniocytes or chorionic villi following quantitative fluorescent polymerase chain reaction for the common aneuploidies, which was also used to exclude maternal cell contamination, and karyotyping or microarray analysis (Figure 1). There was sufficient DNA available with the exception of case 12 (Table 1), in which parents and sibling were sequenced and the diagnosis confirmed in the fetus using Sanger sequencing. Fetuses were excluded if other etiological causes were considered likely, for example intrauterine growth restriction.

bp, basepair; CVS, chorionic villus sampling; gDNA, genomic DNA; GOSH, Great Ormond Street Hospital; USS, ultrasound scan.
Rapid clinical exome sequencing in trios
Parental and fetal DNA was sequenced simultaneously (trio testing) to expedite interpretation of results. A modified “clinical exome” sequencing method was adopted; a custom-designed 20-Mb region of the exome, including all known disease-causing genes was captured (referred to as the “GOSHome”). In this study, with the exception of case 15, we restricted our analysis to a virtual “panel” of 240 genes known to cause skeletal dysplasias and which is updated as knowledge changes (Supplementary Table S1 online).
Library preparation
DNA was extracted from parental whole blood and directly from excess chorionic villi or amniocytes; remaining chorionic villi or amniocytes were cultured and extracted DNA was stored for further testing if required. An Agilent (Agilent Technologies, Santa Clara, CA, USA) SureSelect Focused Exome Plus 1 (GOSHome_v3) enriched a 20-Mb genomic region. Following the manufacturer recommended protocol “SureSelect Target Enrichment System for Illumina (Illumina, Inc., San Diego, CA, USA) Paired-End Multiplexed Sequencing Library (G7530–90000, version B5)” a starting input of 200 ng total genomic DNA was used per sample. After library preparation the trio of libraries were pooled equally and loaded at a final concentration of 1.4 pMol. We performed next-generation sequencing of 150 base pair paired-end reads using a 300-cycle mid output cartridge on the Illumina NextSeq500.
Data analysis
Data were analyzed using our in-house designed Genesis pipeline, which aligned FASTQ files with Burrows–Wheeler Aligner, called variants with FreeBayes v0.9.21, and performed variant annotation with Ensemble Variant Effect Predictor and Alamut-Batch v1.3.1 (Interactive Biosoftware, Rouen, France). Variant interpretation focused on a virtual panel of 240 genes known to cause skeletal dysplasias curated using panelApp (https://panelapp.genomicsengland.co.uk/). The pipeline output was limited to variants within 20 base pairs of the donor and acceptor splice sites of consensus coding sequence exons, and filtering of variants was conducted examining only those with a minor allele frequency of <2% in ExAC (overall frequency), Exome Variant Server, or 1000 Genomes data sets.
Trio analysis
Candidate variants within the gene panel were identified using the variant interpretation platform, Sapientia v1.5.13. This software platform filters variants by Exomiser score (a Sanger Institute–designed Java program that searches for likely pathogenic variants in whole-exome data), population frequency, and mode of inheritance. Previous reports of a variant were determined using HGMD Professional (http://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and locus-specific databases. Variants were classified according to the UK best practice guidelines14 prior to the adoption of the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines6 and then using the American College of Medical Genetics and Genomics guidelines for the more recent cases. Pathogenic, likely pathogenic, and novel variants of uncertain clinical significance were shortlisted as candidate variants.
Confirmation of suspected pathogenic variants
Candidate variants were reviewed by the referring clinical geneticists, fetal medicine experts, and clinical scientists, and any variants considered to be disease-causing were confirmed using Sanger sequencing. Initially confirmation was performed on all members of the maternal–paternal–fetus trio to validate next-generation sequencing data including determining parental inheritance and for additional evidence in de novo cases. Later, in cases where the fetus was homozygous for a pathogenic variant found in both parents, the report was issued without confirmation to expedite return of results to parents. Results were issued to the patient’s clinician in the form of a research diagnostic report.
Results
A total of 19 cases were referred, and after case review 16 were sequenced and a definitive molecular diagnosis was made in 13 (81%) (Table 1). Of the three cases excluded, one pregnancy was terminated before sequencing and investigations were performed postnatally. The second, thought unlikely to have a skeletal dysplasia, was subsequently found to have a chromosomal rearrangement. The third pregnancy is ongoing with growth restriction. Of the three cases sequenced without a definitive diagnosis one (case 1) had a paternally inherited variant, in COL1A1, c.1168G>A p.(Ala390Thr), that is found in ~1% of the population and does not disrupt a GLY residue. This was paternally inherited and because the father was of normal stature, had no signs of OI, and no history of fracturing, this was classified as a variant of unknown significance and not reported. After birth the baby was found to have normal long bones with very mild bowing of the femora detected radiologically. No diagnosis was made after expert genetic review. The second case (case 6) had a heterozygous variant, GDF5 c.902G>A p.(Arg301Gln), which was inherited from the mother who was normal and so too was classified as a variant of unknown significance. The baby was delivered at 29 weeks because of poor growth and decreased fetal movements, with an etiology of placental insufficiency. At 3 months of age she is developing normally and her height remains below the third percentile. In the third (case 15) no pathogenic variants were identified using the skeletal dysplasia panel, but in view of the ultrasound findings indicating possible craniosynostosis, analysis was extended to include a panel including 8 genes associated with craniosynostosis and 123 genes associated with ciliopathies. A single, maternally inherited likely pathogenic variant was identified in the RECQL4 gene that causes Baller–Gerold syndrome, which is compatible with the fetal ultrasound findings. However, as no second pathogenic variant was identified this could not be confirmed as the diagnosis before birth. This pregnancy ended in an intrauterine death and details of the postmortem are awaited.
In all other cases, sequencing results could explain the prenatal phenotype enabling definitive diagnoses to be made. There were four recessive conditions with pathogenic variants inherited from both unaffected parents, six de novo dominant pathogenic variants, and two dominant pathogenic variants inherited maternally (Table 1). In one, (case 7) diagnosis was complicated as the mother had short stature and a maternally inherited 10q26 deletion was demonstrated by microarray analysis. This was initially interpreted as a possible cause for the short stature. A maternally inherited ALPL c.331G>A p.(Ala111Thr) likely pathogenic variant was also identified during sequencing. The mother had no evidence of hypophosphatasia clinically and had normal blood levels of alkaline phosphatase while pregnant but had an increased urinary phosphoethanolamine, raising the possibility of hypophosphatasia. However, biochemical and clinical findings in the newborn baby were consistent with a diagnosis of hypophosphatasia. The second case with an inherited variant (case 8) was referred at 25 weeks’ gestation as the fetus had short, bowed long bones. At genetic consultation, the mother was of short stature, had mild discoloration of her teeth, and a history of two fractures as a child. Recurrent fractures in some relatives were subsequently reported and a possible diagnosis of OI was discussed. Exome sequencing identified the heterozygous pathogenic variant COL1A2 c.2835+1G>A inherited from the mother, confirming a diagnosis of autosomal dominant OI type IV.
The turnaround times from receipt of samples in the laboratory to diagnosis ranged from 11 to 41 days, with times decreasing as we streamlined the multidisciplinary approach and clinical exome-sequencing interpretation protocol.
While in some cases the fetal ultrasound findings alone were of sufficient severity for parents to decide to terminate pregnancies, in others confirmation of molecular diagnosis enabled more accurate prediction of outcome allowing parents more certainty to make a decision to terminate. In at least one case (cases 12) definitive molecular diagnosis enabled the parents to continue the pregnancy. This case was referred at 20 weeks following an ultrasound scan, which showed significant shortening of all long bones, a small chest with normal ossification, and no evidence of bowing or fractures. A previous child had been born with short stature but no definitive diagnosis. The parents were anxious to know if the current pregnancy was an unrelated, de novo, severe skeletal dysplasia or if the fetus had short stature similar to their first child. Amniocentesis had been performed already but there was insufficient DNA available for sequencing. Trio sequencing of the previous child and the parents detected a homozygous frameshift insertion in OBSL1 c.1273dup p.(Thr425Asnfs*40), which was heterozygous in the parents. Sanger sequencing of the remaining amniocyte DNA sample in the current pregnancy showed that the fetus was also homozygous for this pathogenic variant and on this basis the parents elected to continue the pregnancy.
Discussion
Here we describe for the first time the establishment of a rapid diagnostic service for families with the unexpected finding of a possible severe skeletal fetal dysplasia, such that it can be used to influence pregnancy management. It required close collaboration between the fetal medicine teams making the ultrasound diagnosis, and clinical geneticists and clinical scientists both for case selection and interpretation of genomic results. Good communications with local genetic and fetal medicine teams, and sharing of digital images and ultrasound reports enabled inclusion of cases from across the United Kingdom without the need for patients to travel. Knowledge of the genetic diagnosis allowed better prenatal counseling by pediatricians with expertise in managing children with these conditions. Ultrasound alone can suggest a diagnosis and in two cases these findings were of sufficient severity for parents to decide to terminate pregnancies. However, a definitive diagnosis can only be made by molecular analysis or other pathological investigations after birth. In this series, the molecular diagnosis in some cases indicated a less severe prognosis and allowed parents to continue the pregnancy. In some, molecular confirmation of a diagnosis gave increased certainty of poor prognosis, giving parents more confidence to make a decision to terminate an affected pregnancy. With experience, the time to diagnosis fell to less than 2 weeks for the most recent cases, making results more useful in prenatal counseling and parental decision making. A definitive diagnosis was made in 13 of the 16 (81%) cases sequenced, demonstrating the value of careful case selection by multidisciplinary review. In one further case, a molecular diagnosis was thought highly likely following detection of a maternally inherited likely pathogenic variant in a recessive gene in which pathogenic variants are compatible with the phenotype seen. While six arose de novo, we identified four families where the fetus had inherited a pathogenic variant from both parents. In two cases, the condition was inherited from their mothers, both of whom were unaware they had a genetic condition (cases 7 and 8). These cases highlight the need for careful and expert pretest genetic counseling to include the possibility of detecting subtle features of parental carrier status or family history.
Prenatal diagnosis is beneficial for reproductive counseling for conditions leading to neonatal death, such as some severe types of osteogenesis imperfecta. If families decline postmortem, for example for religious reasons, accurate diagnosis can often not be made postnatally. A genetic diagnosis obtained during the pregnancy allows for accurate risk counseling, prenatal diagnosis in future pregnancies, the option of in vitro fertilization with preimplantation genetic diagnosis, or, in the most severe cases, justification for terminating the pregnancy.
However, a sequencing approach to prenatal diagnosis is not without its limitations, which include cost, potential difficulties in interpretation due to the limited phenotyping available prenatally in spite of advances in imaging techniques, limited coverage of disease-causing genes, and ethical issues around the identification of incidental findings if using whole-genome or whole-exome sequencing.15,16,17 There are guidelines for managing these in the postnatal setting,18 but none exist as yet to guide management prenatally where there is the additional ethical challenge given the potential to influence pregnancy decision making based on identifying variants unrelated to the fetal phenotype. In the United States, routine use of whole-genome or whole-exome sequencing for prenatal diagnosis is not recommended outside of the context of clinical trials.19 Furthermore, if using whole-genome or whole-exome sequencing, detection of secondary findings, such as cancer susceptibility genes, denies the unborn child the right not to know if these are disclosed.17,20 Using a trio exome approach and targeted panel analysis, as we have in this study, minimizes this risk of identifying additional findings,8,21 but an important caveat is the incomplete coverage of relevant genes that might result. This may be particularly relevant when one parentally inherited pathogenic variant is identified in a recessive gene that predicts the prenatal phenotype seen as is reported in the literature22,23 and probably demonstrated in case 16.
Simultaneous trio testing increases costs but is also required to facilitate faster filtering and interpretation of gene variants, and confirmation of inheritance to allow accurate discussion of prognosis and recurrence risks in a timely fashion. Cost will become less of an issue as sequencing costs fall24 but for now, particularly in public health settings, it is a barrier that can be minimized by careful multidisciplinary review and case selection, as we have shown in this series.
Accurate interpretation of genome sequences requires detailed knowledge of the phenotype, which in the prenatal setting is not always possible at the time of testing and may be complicated by variable expressivity or evolution of the full phenotype over time. Furthermore, some phenotypes are impossible to determine from prenatal imaging, such as developmental delay, intellectual disability, and behavioral abnormalities. Finally, there is no database for fetal abnormalities equivalent to postnatal dysmorphology databases to help with phenotyping.25,26
Study limitations
This is a small study of highly selected cases. Further study is required to demonstrate applicability to other prenatally detected anomalies to widen eligibility. This study only considered the practical issues of referral, case review, and laboratory aspects and did not formally explore ethical issues or parents’ views, although in our cohort they generally expressed preference of knowing the diagnosis as it allowed emotional preparation, information-gathering, and better preparation for mode and place of delivery. However, even accurate molecular diagnosis can lead to further uncertainty due to incomplete penetrance or variability15 and there is limited information on stakeholder views.16,27 As we consider wider implementation we must explore further stakeholder views and develop guidelines to support implementation.
In conclusion, we have shown that using an expert multidisciplinary approach to case selection and data interpretation, targeted exome sequencing can yield very timely and high diagnostic rates in fetuses presenting with unexpected skeletal abnormalities. In this small series, delivered across the country, these results aided parental counseling and decision making in some cases. Trio testing is required for rapid results, which is expensive, and raises many ethical issues. However, if carefully handled and with strict case selection, this development could prove beneficial in other fetal abnormalities likely to have a genetic etiology. This is a rapidly developing field, with programs such as the 100,000 Genomes Project driving transformation of care for those with rare diseases,28 and health-service providers moving toward implementation of genome sequencing as a first-line investigation. Thus there is an urgent need to develop national guidelines to support timely and appropriate clinical implementation. We believe that this will improve prenatal diagnosis and counseling for parents faced with difficult decisions following the detection of unexpected abnormalities in their unborn baby.
References
Calzolari E, Barisic I, Loane M et al. Epidemiology of multiple congenital anomalies in Europe: a EUROCAT population-based registry study. Birth Defects Res A Clin Mol Teratol 2014;100:270–6.
Fiorentino F, Napoletano S, Caiazzo F et al. Chromosomal microarray analysis as a first-line test in pregnancies with a priori low risk for the detection of submicroscopic chromosomal abnormalities. Eur J Hum Genet 2013;21:725–30.
Nayak SS, Shukla A, Lewis L et al. Clinical utility of fetal autopsy and its impact on genetic counselling. Prenat Diagn 2015;35:685–691.
Chitty LS, Mason S, Barrett AN et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn 2015;35:656–62.
Lewis C, Hill M, Chitty LS. Non-invasive prenatal diagnosis for single gene disorders: experience of patients. Clin Genet 2014;85:336–42.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24.
Wright CF, Fitzgerald TW, Jones WD et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015;385:1305–14.
Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, Chitty LS. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn; e-pub ahead of print 27 June 2017.
Wapner R, Brennan K, Bier L, Wou K, Goldstein D. Whole exome sequencing in the evaluation of fetal structural anomalies: a prospective study of sequential patients. Am J Obstet Gynecol 2017;216(suppl):S5–S6.
Chitty L, Lord J, Rinck G et al. Which fetuses benefit most from exome sequencing? Interim results from the Prenatal Assessment of Genomes and Exomes (PAGE) study. Prenat Diagn 2017;37(suppl 1):3.
David A, Chitty LS, Oepkes D, Gottschalk I, Westgren M, Gotherstrom C. BOOSTB4—A clinical study on pre- and postnatal stem cell transplantation for treatment of osteogenesis imperfect. Prenat Diagn 2016;36 (suppl 1):3–90.
Westgren M, Gotherstrom C. Stem cell transplantation before birth—a realistic option for treatment of osteogenesis imperfecta? Prenat Diagn 2015;35:827–32.
Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016;387:1657–71.
Wallis Y, Payne S, McAnulty C et al, Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf. Accessed 16 March 2018.
Westerfield LE, Stover SR, Mathur VS et al. Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice. Prenat Diagn 2015;35:1022–9.
Kalynchuk EJ, Althouse A, Parker LS, Saller DN Jr, Rajkovic A. Prenatal whole-exome sequencing: parental attitudes. Prenat Diagn 2015;35:1030–6.
Horn R, Parker M. Opening Pandora’s box?: ethical issues in prenatal whole genome and exome sequencing. Prenat Diagn; e-pub ahead of print 10 July 2017.
Green RC, Berg JS, Grody WW et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–74.
Green RC, Berg JS, Berry GT et al. Exploring concordance and discordance for return of incidental findings from clinical sequencing. Genet Med 2012;14:405–10.
Botkin JR, Belmont JW, Berg JS et al. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet 2015;97:6–21.
Abou Tayoun A, Spinner N, Rehm H, Green RC, Bianchi DW. Prenatal DNA sequencing: clinical, counseling, and diagnostic laboratory considerations. Prenat Diagn; e-pub ahead of print 27 March 2017.
Drury S, Williams H, Trump N et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn 2015;35:1010–7.
Mackie FL, Carss KJ, Hillman SC, Hurles ME, Kilby MD. Exome sequencing in fetuses with structural malformations. J Clin Med 2014;3:747–62.
Beckmann JS. Can we afford to sequence every newborn baby’s genome? Hum Mutat 2015;36:283–6.
Landrum MJ, Lee JM, Benson M et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8.
Stenson PD, Mort M, Ball EV et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017;136:1–13.
Quinlan-Jones E, Kilby MD, Greenfield S et al. Prenatal whole exome sequencing: the views of clinicians, scientists, genetic counsellors and patient representatives. Prenat Diagn 2016;36:935–41.
Griffin BH, Chitty LS, Bitner-Glindzicz M. The 100 000 Genomes Project: what it means for paediatrics. Arch Dis Child Educ Pract Ed 2017;102:105–107.
Aptekar L, Gellerman M, Willis AS, Roberts R Whole exome sequencing following the finding of multiple skeletal anomalies on prenatal ultrasound. https://www.integratedgenetics.com/sites/integratedgenetics/files/Whole%20Exome%20Sequencing%20Following%20the%20Finding%20of%20Multiple%20Skeletal%20Anomalies%20on%20Prenatal%20Ultrasound%20.pdf. Accessed 16 March 2018.
Acknowledgments
This project received funding from the National Health Service National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital, and the European Union’s Horizon 2020 research and innovation program under grant agreement no. 681045 that funded the BOOSTB4 study led by Cecilia Götherström. The study sponsors had no role in study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. The views expressed are those of the author and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Chandler, N., Best, S., Hayward, J. et al. Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med 20, 1430–1437 (2018). https://doi.org/10.1038/gim.2018.30
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2018.30
Keywords
This article is cited by
-
What is the power of a genomic multidisciplinary team approach? A systematic review of implementation and sustainability
European Journal of Human Genetics (2024)
-
Using MRI derived observed to expected total lung volume to predict lethality in fetal skeletal dysplasia
Pediatric Radiology (2024)
-
Genetic testing and diagnostic strategies of fetal skeletal dysplasia: a preliminary study in Wuhan, China
Orphanet Journal of Rare Diseases (2023)
-
Experience in prenatal genetic testing and reproductive decision-making for monogenic disorders from a single tertiary care genetics clinic in a low-middle income country
BMC Pregnancy and Childbirth (2023)
-
Chromosomal microarray should be performed for cases of fetal short long bones detected prenatally
Archives of Gynecology and Obstetrics (2021)
