
Abstract
Purpose
Proteus syndrome is a rare mosaic overgrowth disorder that is associated with severe complications. While anecdotal data have suggested that the life span of affected patients is reduced, this has not been measured. Mortality data on rare diseases is critical for assessing treatments and other interventions.
Methods
To address this we used the clinical research records of 64 patients in a longitudinal natural history cohort at the National Institutes of Health to ascertain the data in an organized manner and estimate survival using a Kaplan-Meier approach.
Results
The median age of diagnosis was 19 months. Based on this analysis, there was 25% probability of death by 22 years of age. Ten of the 11 patients who died were younger than 22 years of age, and there was only a single death after this age.
Conclusion
These data quantify the risk of premature death in Proteus syndrome, which can be used to support interventions and trials. Although the risk of death is substantial, the fact that only one patient died after 22 years of age supports anecdotal evidence that the disease process moderates after the end of adolescence. Interventions to reduce mortality should be targeted to the pediatric age range.
Similar content being viewed by others

Introduction
For many rare disorders, it can be challenging to develop appropriate therapeutic approaches because of a dearth of data regarding the morbidity and mortality of the disease. The genetics community is beginning to grapple with this in a systematic manner through the ClinGen actionability process.1 More robust and systematically organized data on rare disorders are necessary to support these, and related efforts. Furthermore, the development of therapeutic approaches for these diseases must be based on objective data on the natural history of the disease, both as a motivating factor and as a metric against which the risks of treatment can be properly balanced. Proteus syndrome is a rare, somatic overgrowth disorder2 caused by a mosaic3 c.49G>A p.(E17K) mutation in AKT1, which encodes the V-Akt murine thymoma viral oncogene homolog 1, a key effector of growth signaling pathways. The disorder has remarkable pleiotropy, potentially affecting essentially any organ system. The most common manifestations are skeletal overgrowth, nevi, vascular malformations, dysregulated adipose growth, CNS anomalies, and others. For a number of years there was substantial diagnostic confusion regarding this disease, but this has declined with both the application of specific clinical diagnostic criteria4 and molecular testing for the causative mutation in affected tissues.
In spite of these advances, several challenges remain, including the management of the overgrowth and a number of medical consequences that can be life-threatening, including deep-vein thrombosis/pulmonary embolism,5, 6, 7 an apparently increased susceptibility to cancer,8, 9 and others.10 It is critical to accurately assess the morbidity and mortality of Proteus syndrome to support clinical decision making and focus research efforts on disorders that substantially reduce life span. To address the question of mortality and frame it in context of age at diagnosis, we evaluated a large cohort of affected patients for these two attributes.
Materials and methods
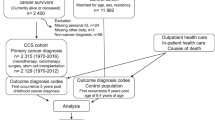
The data were abstracted from clinical research charts of patients with Proteus syndrome who participated in protocol 94-HG-0132 (NCT 00001403), “The Phenotype and Etiology of Proteus Syndrome and Related Overgrowth Disorders” at the National Human Genome Research Institute (NHGRI). The study was reviewed and approved by the NHGRI institutional review board. Charts comprised clinical data obtained through evaluations at the National Institutes of Health (NIH) or outside institutions and were primarily in paper format. Patients with insufficient clinical data to allow accurate data abstraction were omitted from this analysis. The NIH Clinical Center electronic medical records system (CRIS) was used to update or confirm the patients’ date of last contact or death. Two primary reviewers with medical knowledge (J.Z. and L.H.) reviewed the charts of all patients, abstracted values for each variable independently, and adjudicated differences via additional chart reviews or by discussing with one of the study team (J.C.S.).
Data abstraction
Information on the following six variables was identified, abstracted and entered into a spreadsheet. 1. Patient medical record number: abstracted directly from the charts. 2. Date of birth: abstracted directly from the charts. 3. Gender: abstracted directly from the charts. 4. Age/date of death (if applicable): abstracted from the charts, CRIS, or correspondence with study staff. 5. Age of meeting clinical diagnosis criteria or first diagnosis for Proteus syndrome (whichever came first): revised clinical diagnostic criteria4 were used as guidance for identifying the age of meeting clinical diagnosis criteria. Because the identification of values for this variable is subjective, the primary reviewers and J.C.S. held several meetings to discuss the interpretation of these diagnostic criteria. Ten example cases were reviewed together and the primary reviewers then conducted another round of review for consistency. Specifically, the information on this variable was identified in physicians’ notes, letters, or laboratory results consistent with either the interpreted clinical diagnostic criteria or the first diagnosis time, whichever came first. Each reviewer also made independent notes indicating the basis of the decision about the age/date for each patient. The rationale for selecting the earliest age either of meeting the clinical diagnostic criteria or at the first diagnosis is that most patients had already met the clinical diagnostic criteria before a diagnosis was formally made. In the interest of concision, we refer below to the age of meeting clinical diagnostic criteria or first diagnosis as the “disease inception age.” The sixth variable is date of last contact: the most recent of the following three dates was selected: (i) last contact date, information identified from the paper charts; (ii) last contact date, information in the CRIS system; or (iii) last contact date, information documented by email correspondence.
Three Kaplan-Meier survival curves were plotted for these patients showing (i) years from birth to disease inception; (ii) years from birth to death; and (iii) years from disease inception to death. SAS 9.4 was used to generate the Kaplan-Meier curves and all descriptive statistics.
Results
A total of 64 patients with Proteus syndrome were identified in this study; 28 (43.8%) were female and 36 were male (56.3%). The mean and median disease inception ages were 3.2 and 1.6 years old, respectively. In all, 11 of the 64 patients (17.2%) had died at the time of chart review. Of the 11 deaths, 10 patients were younger than 21.8 years of age at the time of death. The mean and median ages at time of death among the 11 patients were 14.4 (±11.3) and 15.1 years, respectively. Of the 53 patients who were still living at the date of last contact, 36 (67.9%) were younger than 21 years old. The median value for date of death or date of last contact by the NHGRI research team since a patient’s disease diagnosis was 11.5 years. The Kaplan-Meier survival curve for the outcome (e.g., death) shows, as a function of time, the probability of having the event (in this case, death). The survival curve is also decorated with vertical tick marks indicating the ages of last contact for patients who did not die during the study period.
We first evaluated the age of diagnosis. For this evaluation, the Kaplan-Meier graph can be best considered as the fraction of patients who are undiagnosed (Figure 1). Because none of the participants in this study were diagnosed antenatally (and we are unaware of a prenatal diagnosis of proven Proteus syndrome), 100% were undiagnosed at age 0. The median age at diagnosis was about 19 months of age, and 75% were diagnosed by age 3.2 years. We next evaluated age at death (Figure 2). The Kaplan-Meier curve (Figure 2) suggests that a majority of patients (74.2%) have a chance to survive into adulthood. However, mortality was significant, with a relatively consistent rate of death from infancy through the beginning of the third decade. No patients in the study died after approximately 22 years of age. The results were not significantly different when the interval was instead analyzed from age at diagnosis to age at death (Supplementary Figure S1 online). The causes of death, when known, are reported in Supplementary Table S1.

The Kaplan-Meier curve for the disease inception age. Note that no patients were censored in this figure, because all patients necessarily had a disease inception age. In this plot, the y axis can be considered the fraction of patients as yet undiagnosed.

The Kaplan-Meier curve for age at death, with the interval in years from birth. From this analysis, about 75% of patients are predicted to be still living at the age of 21.8 years.
Discussion
An understanding of the natural history of rare genetic diseases is critical to support efforts to better diagnose and treat these diseases. Recognition of the severity of diseases can be measured by mortality, although mortality alone is an incomplete measure as the morbidity of living patients is equally important. For many diseases, we have only anecdotal data or case reports from which it is extremely difficult to accurately estimate morbidity and mortality. The incidence of Proteus syndrome is probably on the order of 1/10,000,000 births—one of the rarest known disorders. Given also the well-recognized diagnostic confusion of Proteus syndrome with other disorders,4, 11, 12 it is difficult to estimate morbidity and mortality. While our group and others have published observations of morbidity and mortality from this disorder and were well aware of its seriousness, no objective measure of mortality from any cohort was available. Although the cohort here is relatively small (64 patients), it is the largest confirmed cohort of patients with Proteus syndrome.
The utility of the NHGRI Proteus syndrome cohort is that the patients are evaluated in detail and the diagnoses are confirmed. All of these patients meet clinical criteria4 and nearly all patients for whom an appropriate tissue sample was available (48 of 49 patients; 98%) have had molecular confirmation of the AKT1 c.49G>A p.(E17K) mutation in one or more tissues.3 The analysis of this cohort shows that the median age of diagnosis was about 19 months. This median age of diagnosis is plausible, seeing that the age of onset often correlates with the rapid phase of overgrowth which typically occurs between 6 months and 2 years of age.4 We suggest that this surge of asymmetric, distorting overgrowth in infants and toddlers triggers parental concern, which leads to a diagnosis. While the average age at diagnosis is in the toddler phase, it is concerning that for many patients, this takes much longer, the latest being 24 years of age. Our unpublished data show that many of these later-diagnosed patients suffered longstanding emotional and psychosocial challenges from being misdiagnosed or undiagnosed. We hope that increased recognition of the disorder, improved application of the published clinical criteria, and use of molecular genetic testing will improve this.
We next quantitated the mortality of Proteus syndrome. We and others have documented causes of death,12 including venous thromboembolism,5, 6, 7 cancer,8, 9 and others.10 While it was clear to us that the disease was serious and that young people were dying of this disease, it was difficult to quantitate. By systematically reviewing the records of our cohort we have been able to solidify this impression of early mortality and show that nearly a quarter of the patients enrolled in this protocol died before they reached their mid-20s. There was a relatively consistent rate of deaths starting in infancy and continuing until the late teens. The mortality rate was lower among patients who had survived until their early 20s.
These data have several limitations. First, we did not systematically recontact every individual in this cohort to determine if they were alive or if they had died after our last contact with them. This is reflected in the large number of censored data points on Figure 2. Second, our study may have been biased in respect of ascertainment. One of the more likely biases would be toward greater severity as patients and parents of patients (where children are affected) may be more likely to enroll in our study if they are more severely affected. However, this may be counterbalanced by the fact that a number of very severely affected patients, including one who died recently,13 were so severely affected that it was not practical for them to travel to the NIH to participate in the study. This bias would reduce the number of the most severely affected patients in the study. Finally, the cohort size of 64, while impressive for an extremely rare disorder, is in a statistical sense not a large population, and the precision of our estimates of both age of diagnosis and age at death is limited. Of note, nine patients with confirmed AKT1 c.49G>A p.(E17K) mutations found in our laboratory were excluded from this analysis because insufficient clinical data were present to confirm satisfaction of clinical diagnostic criteria or another of the variables used in the analysis.
In summary, these data confirm the clinical impression that Proteus syndrome is a severe, typically pediatric-onset disorder. Children and young adults are susceptible to premature death and clinicians and researchers need to address these challenges to longevity through basic research and clinical studies and therapeutic trials of primary and symptomatic treatments in the pediatric age range.
References
Hunter JE, Irving SA, Biesecker LG et al, A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med 2016;18:1258–1268.
Biesecker LG . The multifaceted challenges of Proteus syndrome. JAMA 2001;285:2240–2243.
Lindhurst MJ, Sapp JC, Teer JK et al, A mosaic activating mutation in AKT1 associated with the Proteus syndrome. New Engl J Med 2011;365:611–619.
Biesecker LG . The challenges of Proteus syndrome: Diagnosis and management. Eur J Hum Genet 2006;14:1151–1157.
Slavotinek AM, Vacha SJ, Peters KF & Biesecker LG . Sudden death caused by pulmonary thromboembolism in proteus syndrome. Clin Genet 2000;58:386–389.
Eberhard DA . Two-year-old boy with Proteus syndrome and fatal pulmonary thromboembolism. Pediatr Pathol 1994;14:771–779.
Papadimitriou L, Kavalieratos CS . Proteus syndrome and sudden cardiac arrest; beware of pulmonary embolism!. Resuscitation 2002;54:313–314.
Gordon PL, Wilroy RS, Lasater OE, Cohen MM Jr . Neoplasms in proteus syndrome. Am J Med Genet 1995;57:74–78.
Iqbal J, He G, Biesecker LG et al, Morphological characterization of the breast in proteus syndrome complicated by ductal carcinoma in situ. Ann Clin Lab Sci 2006;36:469–474.
Cohen MM Jr . Causes of premature death in Proteus syndrome. Am J Med Genet 2001;101:1–3.
Biesecker LG, Happle R, Mulliken JB et al, Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet 1999;84:389–395.
Turner JT, Cohen MM Jr, Biesecker LG . Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am J Med Genet 2004;130A:111–122.
Doucet ME, Bloomhardt HM, Moroz K, Lindhurst MJ, Biesecker LG . Lack of mutation-histopathology correlation in a patient with Proteus syndrome. Am J Med Genet A 2016;170:1422–1432.
Acknowledgements
This secondary data analysis study was supported by funding from ArQule, Inc. The primary natural history study and the efforts of all the NIH coauthors were supported by NIH grants ZIA HG200328 11 and ZIA HG200388 03 from the Intramural Research Program of the National Human Genome Research Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
L.G.B. receives royalties from Genentech Inc. and honoraria from Wiley-Blackwell Inc. and is an uncompensated adviser to the Illumina Corp. B.S. and D.F. are employees and hold equity in ArQule, Inc. The other authors declare no conflict of interest.
Additional information
Supplementary material is linked to the online version of the paper at
Rights and permissions
About this article

Cite this article
Sapp, J., Hu, L., Zhao, J. et al. Quantifying survival in patients with Proteus syndrome. Genet Med 19, 1376–1379 (2017). https://doi.org/10.1038/gim.2017.65
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.65
Keywords
This article is cited by
-
Development of the Clinical Gestalt Assessment: a visual clinical global impression scale for Proteus syndrome
Orphanet Journal of Rare Diseases (2022)
-
A case report of Proteus syndrome (PS)
BMC Medical Genetics (2020)

{kind=link}
{kind=link}
{kind=link}