
Abstract
Purpose
Smooth muscle dysfunction syndrome (SMDS) due to heterozygous ACTA2 arginine 179 alterations is characterized by patent ductus arteriosus, vasculopathy (aneurysm and occlusive lesions), pulmonary arterial hypertension, and other complications in smooth muscle–dependent organs. We sought to define the clinical history of SMDS to develop recommendations for evaluation and management.
Methods
Medical records of 33 patients with SMDS (median age 12 years) were abstracted and analyzed.
Results
All patients had congenital mydriasis and related pupillary abnormalities at birth and presented in infancy with a patent ductus arteriosus or aortopulmonary window. Patients had cerebrovascular disease characterized by small vessel disease (hyperintense periventricular white matter lesions; 95%), intracranial artery stenosis (77%), ischemic strokes (27%), and seizures (18%). Twelve (36%) patients had thoracic aortic aneurysm repair or dissection at median age of 14 years and aortic disease was fully penetrant by the age of 25 years. Three (9%) patients had axillary artery aneurysms complicated by thromboembolic episodes. Nine patients died between the ages of 0.5 and 32 years due to aortic, pulmonary, or stroke complications, or unknown causes.
Conclusion
Based on these data, recommendations are provided for the surveillance and management of SMDS to help prevent early-onset life-threatening complications.
Similar content being viewed by others

Introduction
Smooth muscle dysfunction syndrome (SMDS, MIM 613834) presents with a recognizable pattern of complications, including congenital mydriasis, patent ductus arteriosus (PDA), pulmonary arterial hypertension, aortic and other arterial aneurysms, moyamoya-like cerebrovascular disease, intestinal hypoperistalsis and malrotation, and hypotonic bladder.1 It is caused by heterozygous mutations of the ACTA2 altering arginine 179, most commonly p.Arg179His. With a single exception, all cases are due to de novo mutations. Seven SMDS cases were initially reported,1 followed by reports of cases with similar presentation and additional complications often resulting in poor health outcomes and early deaths.2,3,4,5,6,7,8,9,10,11,12,13
The clinical evolution of the recently identified SMDS has not been described and guidelines for evaluation and medical management are lacking. Therefore, we sought to describe the clinical history and outcomes of SMDS patients. These data are used as a basis to propose guidelines for clinical evaluation and management of patients with SMDS and direct future therapeutic trials.
Materials and methods
The study was approved by the institutional review board of the University of Texas Health Science Center at Houston, and informed consent was obtained from the participants or parents. Outside investigators provided de-identified patient data after obtaining approval from their respective institutional review boards and consent from the patient or family.
Thirty-three patients with confirmed mutations of the ACTA2 Arg179 from North America, Europe, Asia, and Australia were enrolled from 2009 to 2017. Medical information were collected through review of available medical records (Supplemental Information online). All reported data reflected the patient’s diagnosed medical conditions and treatment at the last clinical evaluation.
Data were analyzed using STATA/IC version 12.1 (College Station, TX). Categorical variables were summarized by frequency and percentage and continuous variables by median, interquartile range (IQR), minimum/maximum values. Failure functions of clinical endpoints (i.e., aortic aneurysm repair or dissection, stroke, or death) were calculated using the Kaplan–Meier method.
Results
Characteristics of the study population
Thirty-three patients with ACTA2 Arg179His (24), Arg179Cys (7), Arg179Leu (1), or Arg179Ser (1) mutation were included (including seven previously reported cases1). Except for one, all patients had de novo mutations. Median age at diagnosis by genetic testing was 11 years (IQR 15, 0.03–36). Twenty-one (64%) were females and only 12 were males (36%). By race or ethnicity, 26 were of European descent, 5 were Asian, 1 was African American, and 1 was Hispanic/Latino.
Cardiac, aortic, and peripheral arterial complications
All patients had a patent ductus arteriosus (91%) or an aortopulmonary window (APW; 9%) diagnosed during infancy (Supplemental Information). PDAs were noted to be large or aneurysmal and often associated with pulmonary arterial hypertension. In two patients with PDA, the lesion was noted to be large or broad-based with an APW physiology. Twenty-nine patients with available surgical information had ligation or patch closure of the PDA or APW; 25% of patients underwent this surgery within the first month of life, 50% within 2 months, and 90% within 12 months (Table 1). Four patients required re-operations due to residual shunting. In seven patients, the PDA or APW was present in the setting of aortic arch obstruction or aortic coarctation. Eleven patients also had atrial septal defects and one had a ventricular septal defect.
Twelve patients (36%) had had an aortic event, defined as an elective aortic aneurysm repair or dissection, at median age of 14 years (IQR 7, 10–25; Table 1). Cumulative risk of aortic event was 100% by the age of 25 years (Figure 1a). In seven patients, this first event was elective repair of an ascending aortic aneurysm, involving the aortic root and arch. Two children, ages 10 and 12 years, had repair at 42 mm (z-score 7.8) and 49 mm (z-score 8.1), respectively, and two adults, age 24 and 25 years, had repair at 55 mm (z-score not available) and 58 mm (z-score 8.1), respectively. Three patients had valve-replacing aortic root replacement, three had valve-sparing aortic root replacement, and one had an ascending aortic replacement only. Four of these patients (age 12–25 years) had concomitant total aortic arch replacement.

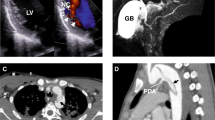
Cumulative risk of first aortic event, stroke, and death associated with smooth muscle dysfunction syndrome. CI, confidence interval.
Two patients had type A aortic dissections at 17 and 24 years of age, and three patients had type B aortic dissections (DeBakey type IIIA in one and IIIB in two patients), all at the age of 14 years. One patient with type A dissection was found to have a chronic dissection of the ascending aorta when she presented for repair of a 53-mm ascending aortic aneurysm. One patient had a type B dissection (#18) when he was being monitored for a 39-mm ascending and 41-mm descending thoracic aortic dilation. The second patient (#20) developed a type B dissection within 24 hours after undergoing a second stage right cerebral revascularization procedure; previous imaging showed the proximal descending aorta measured 35 mm and the ascending aorta was 63 mm. The dissection was complicated by occlusion of the superior mesenteric artery and small bowel ischemia. A stent was placed in the superior mesenteric artery and the aortic dissection was medically managed. The patient died 10 days later; autopsy was not performed. The third patient (#22) with type B dissection was medically managed and died suddenly 3 years later of unknown cause after being lost to follow-up.14
Of these 12 patients with prior aortic events, four had a subsequent aortic aneurysm repair or dissection. One patient (#31) developed rapid enlargement of her suprarenal thoracoabdominal aorta from 44 to 50 mm and underwent open thoracoabdominal repair. The second patient (#32) was being monitored for an aortic arch aneurysm (48 mm on last measurement) due to previous strokes with sequela of left hemiparesis and died of a ruptured type B dissection at the age of 32 years. The third patient (#29) underwent surgery for aneurysms of the arch and abdominal aorta at the age of 31 years, and died from postoperative complications. The fourth patient (#33) underwent open repair of the distal aorta (details not available). Of the patients who had previous elective aortic aneurysm repair and no other aortic events (n = 4), three have thoracoabdominal aortic dilation (Figure 2).

Reconstructed magnetic resonance angiogram images in a 13-year-old patient with ACTA2 Arg179His alteration. (Top) Dilation of the aortic root and ascending aorta and bilateral subclavian and axillary artery aneurysms. (Bottom) Dilation of the descending and thoracoabdominal aorta.
The patients with no history of an aortic event (n = 21) were significantly younger (median age at last follow-up was 5 years, range 0.1–20.5) compared with the median age at first aortic event in the 12 patients above (P = 0.0007). Of 19 patients who had imaging of the proximal thoracic aorta, 17 (90%) were noted to have dilation of the aortic root and ascending aorta (n = 9) or the ascending aorta sparing the aortic root (n = 8). Median maximum diameter of this segment was 27 mm (range 18–68) at last follow-up. Of 14 patients who had evaluation of the aortic arch and proximal descending thoracic aorta, four had a dilated aortic arch and none had a dilated descending aorta. Seven patients who had evaluation of the distal thoracic and abdominal aorta did not have dilation of these segments.
Eighteen (54%) patients had peripheral arterial dilation at median age of 8 years. Sites of arterial dilation include the proximal internal carotid artery (44%), common carotid (22%), brachiocephalic (24%), subclavian (12%), axillary (9%), and external/internal iliac arteries (3%). None of the patients had other arterial dissection or rupture. Three patients with axillary artery aneurysms required surgical repair due to acute thrombosis (Table 1; Supplemental Information). One patient (#27) has a chronic arteriovenous malformation in the right forearm. No coronary artery disease has been reported.
Low blood pressure was common in SMDS patients. Median systolic blood pressure was 101 mmHg (IQR 15, 58–108, n = 14) among young patients age ≤18 years and was 113 (IQR 41, 80–123, n = 7) among adult patients. Median diastolic blood pressure was 50 mmHg (IQR 11, 23–88) among young patients and 54 mmHg (IQR 22, 40–75) among adults.
Of 29 patients with detailed records of medications, medications taken at the last clinical evaluation are shown in Supplemental Figure 1.
Cerebrovascular complications
Patients with SMDS have distinct cerebrovascular disease, including dilation of the proximal carotid arteries, widespread occlusive arteriopathy characterized by moyamoya-like stenotic/occlusive lesions in the distal internal carotid arteries and its branches and the posterior circulation, absence of basal collateral vessels, abnormally straight course of the intracranial arteries, and periventricular and deep white matter signal changes on magnetic resonance imaging (MRI).3 Intracranial artery stenosis or occlusion was present in 23 (77%) patients at median age of 8 years (IQR 10) and as early as 5 days of age. In 16 (53%) patients, stenosis involved more than one artery, commonly the distal internal carotid (53%), cerebral (60%), vertebral (20%), and basilar artery (17%). The majority of patients (95%) had T2-weighted hyperintense periventricular white matter changes on brain MRI.
Eight females and one male had cerebral ischemic infarction at median age of 4 years (IQR 7; Figure 1b). Seven of these patients presented before 10 years, the youngest of whom was 2 days old with bilateral watershed and basal ganglia infarction. The other two patients had had previous composite aortic valve graft replacement (Table 1).15
Five patients underwent cerebral vessel revascularization involving one (n = 1) or both (n = 4) sides and using indirect encephaloduroarteriosynangiosis in four patients and both direct and indirect extracranial to intracranial arterial bypass procedures in one patient (Table 1). Pre- and postoperative cerebral imaging was available for three patients that showed effective collateralization of hypoperfused areas in the middle cerebral artery territory, predominantly provided by neoform vessels arising from the middle meningeal artery (Figure 3). One patient died 3 years later of a stroke, two others died of unknown causes, and the other two patients continued to have transient ischemic symptoms.

Representative images before (top row) and 1 year after (bottom row) indirect revascularization surgery (dural inversion). Angiographic images show multiple abnormalities including straightening of cerebral arteries, dilation of the petrous and cavernous portions followed by severe stenosis of the terminal segment of the right (R) and left (L) internal carotid arteries (ICAs), severe stenosis of the middle and anterior cerebral arteries (with distal R M1 occlusion), and areas of parenchymal hypoperfusion (dashed circles). Follow-up angiogram a year after surgery showed new collateral vessels arising from the middle meningeal arteries (arrows) in the watershed regions of hypoperfusion, with concomitant enlargement of the parent middle meningeal vessel indicating compensatory increased flow, and retrograde flow through the R middle cerebral artery (arrowhead) supporting hemodynamically significant supply from the donor vessels. Magnetic resonance fluid-attenuated inversion recovery images illustrate white matter injury and ischemic infarcts at both time points. ECA, external carotid artery.
Six patients (18%), age 1–19 years, had seizures (Supplemental Information). Four patients had structural brain abnormalities, most commonly malformation of the corpus callosum described as thinning or foreshortening. One had colpocephaly and another had deficiency in development of the rostrum. Areas of polymicrogyria were noted in two patients. Six patients (19%) had mild developmental delay and two (7%) had learning difficulties.
Pulmonary complications
Sixteen patients (48%) had a history of pulmonary arterial hypertension and one patient (#27) required lung transplant at the age of 18 months. Patient #2 presented with respiratory distress at birth and a PDA was diagnosed, but not repaired. He developed severe pulmonary arterial hypertension and significant parenchymal disease, and died at 6 months of age after multiple pulmonary hypertensive crises.
Eleven patients (33%) were diagnosed with chronic lung disease and five other patients with chronic asthma. Seventeen patients were noted to have dilation of the main pulmonary artery and/or its branches. Patient #31 underwent plication of the main pulmonary artery concomitant with aortic root, ascending and arch replacement. This patient had chronic pulmonary disease requiring oxygen supplementation and multiple episodes of right heart failure starting at the age of 29 years, and died at the age of 31 years after hospitalization for pneumonia complicated by aspiration.
Other complications
All patients had pupillary abnormalities described in the medical records as fixed or nonreactive pupils (congenital mydriasis), iris hypoplasia, and/or aniridia or partial aniridia. Seven patients had retinal artery tortuosity and one was noted to have small retinal and vitreous hemorrhages. One adult patient had retinal detachment.
The frequencies of gastrointestinal and urogenital complications were variable (Table 2).
Pregnancy
One pregnancy was observed in a 26-year-old G4P0030 patient who presented for genetic evaluation at 24 weeks of gestation due to a history of PDA repair, ascending aortic aneurysm repair with mechanical aortic valve replacement, and aortic coarctation repair, and was later diagnosed with SMDS during pregnancy. She had an unremarkable brain magnetic resonance angiogram (MRA) and no distal aortic enlargement. Fetal ultrasound at 32 weeks of gestation showed mild dilation of the ascending aorta, an enlarged fetal bladder, and hydronephrosis; testing after birth confirmed the child had inherited the ACTA2 alteration. Planned Cesarean section at 37 weeks gestation demonstrated an atonic uterus and friable tissues during the procedure.
Causes of deaths
Figure 1c shows the risk of death is high among patients under the age of 18 and over the age of 30 years. Nine (27%) of the patients died of complications (median age 17 years, IQR 19, 0.5–32), with three being diagnosed with SMDS after death (Table 1). Deaths were due to confirmed or probable aortic dissection (n = 3), postoperative complications of aortic surgery (1), stroke (1), or pulmonary complications (3). One patient (#16) died of undetermined causes despite an autopsy. The oldest living patient in this cohort was 37 years old.
Discussion
Our findings confirm the cardinal features of SMDS are PDA or APW, thoracic aortic aneurysm and dissection, thoracoabdominal aortic aneurysm, peripheral artery aneurysms, cerebrovascular disease, retinal vessel tortuosity, congenital mydriasis, pulmonary artery hypertension, chronic lung disease, hypotonic bladder, undescended testes, atonic gravid uterus, gut malrotation, hypoperistalsis, and gall bladder sludge or cholelithiasis. Congenital mydriasis and related pupillary abnormalities and PDA or APW were fully penetrant in infancy. Thoracic aortic and cerebrovascular disease were fully penetrant with age. Penetrance of the other complications were variable.
As all patients had a PDA or APW and congenital mydriasis or related pupil abnormalities, newborns with these complications with or without the other systemic complications should raise the suspicion for SMDS and be referred to a geneticist for evaluation and undergo genetic testing for an ACTA2 alteration. Complications can present early in life and require evaluation by multiple specialists (Table 3). Multidisciplinary care of these patients is recommended because medications used to treat complications in one system may aggravate disease in another.
Surveillance and management of SMDS complications
PDA and APW
PDAs were large, and the majority of patients required surgical correction in the first year of life. Untreated or late treatment of PDA is associated with progressive respiratory deterioration, prolonged ventilator support, and death.6,12 Surgical correction of the PDA should be considered in infants with SMDS, because delaying this surgery has the potential to escalate the pulmonary arterial hypertension. In patients who are not candidates for definitive closure of the PDA due to high pulmonary vascular resistance, partial closure by surgery or catheterization may be considered, followed by management with pulmonary vasodilating agents and complete closure when the vascular resistance decreases.16
Complications of the aorta and its branches
Ascending aortic dilation was evident even in utero although no aortic events occurred until the age of 10 years for ascending aortic aneurysm repair and 14 years for aortic dissection (type B). Based on these findings and a previous study,11 the aortic root, ascending aorta, and arch progressively enlarge and may require repair between late childhood and early adulthood.
Evaluation of the entire aorta at the time of diagnosis is recommended. Follow-up imaging should be performed by echocardiography (if the root and ascending aorta are adequately visualized) or cardiac MRI/MRA with cardiac-gated sequences for the aortic root. Based on the earliest clinically significant dilation of the descending thoracic and abdominal aorta observed in this cohort, surveillance of these segments should be started at 10 years of age or earlier if abnormalities were noted on the initial scan.
Patients may benefit from drug treatments that have been shown to slow aortic growth. In patients with Marfan syndrome, the standard of care is β-blockade therapy.17Results of a recent randomized clinical trial have shown that β-blocker and losartan are equally effective in reducing the rate of aortic root dilation in patients with Marfan syndrome.18 There is limited data on the effectiveness of other drug treatments for aortic disease, and no therapies have been specifically tested in individuals with ACTA2 mutations. Given the hypotension and steno-occlusive cerebrovascular disease in these patients, low doses of these medications should be used initially with close monitoring of blood pressure and symptoms. Exercise restrictions are recommended, including avoidance of isometric exercises and contact sports.
The risks and benefits of elective surgery of the thoracic or thoracoabdominal aorta in light of the patient’s comorbid conditions (i.e., cardiac, cerebrovascular, pulmonary disease) should be carefully considered. Neurosurgical evaluation prior to surgical intervention may be required to ensure adequacy of cerebral perfusion while on cardiopulmonary bypass. When possible, elective aortic root repair should be delayed in children until the annulus can accommodate an adult-sized graft. Valve-sparing procedures should be performed to avoid the increased risk of thromboembolism associated with mechanical aortic valves. Concomitant aortic arch replacement should be considered because the data collected here indicate that the arch will enlarge over time. Placement of an elephant trunk in preparation for a staged procedure should be considered because the descending or thoracoabdominal aorta often enlarge over time. Given that a third of patients with other ACTA2 mutations experienced type A dissections when the maximum aortic diameter was <50 mm,19 elective surgical repair should be discussed when the aortic diameter reaches 45 mm. Limited data on type B dissections indicate that these were not preceded by significant dilation and occurred at younger ages compared with type A dissections. Puberty appears to be a high-risk period for type B dissections in SMDS patients and occurred only in males in this cohort. Rate of growth of descending thoracic and thoracoabdominal aortic aneurysms should be closely monitored because there was rapid enlargement in patients. In patients who are suitable candidates for surgery, descending or thoracoabdominal aortic procedures may be electively performed in surgical centers with experience. Patients with descending thoracic aortic dissections should be closely monitored, and treatment individualized. Given the poor outcomes, i.e., aortic rupture and sudden death, in SMDS patients who had a thoracic aortic aneurysm greater than 45 mm or a descending aortic dissection, elective open or endovascular aortic repair should be considered in these situations when possible.
Peripheral artery aneurysms may present with symptoms due to local compression of nearby structures, limb ischemia with thromboembolism, or rupture, although arterial dissection or rupture has not been observed with SMDS. Baseline evaluation by ultrasound, computed tomography, or MRA is recommended to screen for upper-extremity artery aneurysms or malformations and assess the size and presence of thrombus. Annual surveillance by ultrasound of asymptomatic aneurysms should be performed. Surgical repair of symptomatic, thrombotic, or large aneurysms (>20 mm in diameter) should be considered.20 Repair with aneurysm resection and interposition grafting with an autologous vein or prosthesis is recommended. Given the poor performance of stent grafts (high mobility, risk of endoleak due to the rich collaterals of the axillary artery), these should be used only as a temporizing measure in an unstable patient or a definitive measure in patients who are unable to tolerate open repair. The hypotension present in SMDS patients potentially increases the risk of anesthesia and should be taken into account during surgical procedures.
Neurologic complications
Cerebrovascular disease, including stenosis or hypoplasia of arteries and white matter changes, are present as early as the neonatal period in this study and previous reports.3,7,8,21 Patients have a high risk of vaso-occlusive strokes and seizures, the majority of which were observed in neonates and young children. It is not clear whether the seizures were provoked by small vessel vasculopathy or hypoxic–ischemic injury. However, chronic hypotension in the setting of cerebral vasculopathy compromising blood flow and autoregulation to the brain are significant risk factors for cerebral damage in these patients.
A baseline brain MRI and head and neck MRA should be performed to evaluate the burden of white matter injury, ischemic–hypoperfusive events, and degree of stenosis, and to determine future risk of stroke and management to prevent neurologic complications. Because radiation is a known risk factor for moyamoya disease,22,23 MRI is recommended instead of computed tomography. MRI with sedation should be performed in experienced facilities with adequate equipment and well-trained personnel.24 When possible, MRI should be scheduled and coordinated among specialties to maximize the extent of imaging and reduce the child’s exposure to sedation.
Although disease progression and optimal screening intervals have not been established, periodic screening for cerebrovascular disease should be considered as part of the patient’s surveillance. Transcranial Doppler may be useful in monitoring patients with SMDS, but additional studies are needed to examine its utility in predicting risk of stroke in these patients. Conventional catheter angiogram should be reserved for patients with focal neurologic symptoms or evidence on MRA or transcranial Doppler of critical or progressive narrowing of the cerebral arteries.
Patients who had seizures were managed with standard anticonvulsant therapy. Treatment with aspirin may be considered in asymptomatic patients with evidence of cerebral vasculopathy for the prevention of a first stroke.25 Young children on aspirin should be monitored given the risk of Reye syndrome. Patients who had prior aortic root repair with mechanical valve replacement or thromboembolic events should be monitored and sufficiently anticoagulated. Hemorrhagic complications while on anticoagulation therapy have not been reported in SMDS patients. Patients, especially young children, should be closely monitored during illness resulting in low oxygen saturation and acute infections with fever increasing cerebral metabolic demand. Any sudden change in neurologic function should be urgently evaluated. Patients should be counseled about staying sufficiently hydrated and taking measures such as sitting or lying down with the legs elevated if sudden dizziness or lightheadedness occurs.
Although surgical revascularization is widely used in patients with symptomatic moyamoya disease,26 a higher rate of postoperative ischemic stroke was observed in patients with SMDS compared with those with moyamoya disease.3 Limited data on three patients who had indirect cerebral revascularization showed formation of collateral vessels and improved cerebral perfusion, but no overall reduction in risk of stroke or transient ischemic symptoms.
Finally, a small number of patients were noted to have mild developmental delay or learning disability, which may be attributed to cerebral hypoperfusion or infarction, other medical complications, or frequent hospitalizations early in life. Routine cognitive and psychological evaluation should be performed to assess the need for further neurologic evaluation, rehabilitation, and adjustments at school or home.
Pulmonary complications
Pulmonary arterial hypertension in the setting of PDA or APW have been reported in these patients, as well as asthma and poorly characterized chronic lung disease. The cause of the chronic lung disease has not been determined but may be due to a developmental defect in the lung parenchyma predisposing to emphysema. Only one patient required lung transplant due to pulmonary arterial hypertension. Other patients required supplemental oxygen early in life and one adult required permanent oxygen supplementation. In addition, treatment with respiratory tract agents such as bronchodilator or anti-inflammatory agents was prevalent across all patient age groups.
Gastrointestinal complications
Gut malrotation or immotility presents early in life and may require a surgical procedure to correct the intestinal malrotation. Feeding difficulties and failure to thrive may require gastric or nasogastric tube feeding to improve nutrition. Chronic constipation due to gut immotility was treated with laxatives. Patients who develop gall bladder sludge or stones may require gall bladder resection. Hepatic dysfunction was previously reported in two SMDS patients3 but was not observed in this study.
Urogenital complications
Some of the urogenital complications such as enlarged bladder and hydronephrosis were detected in utero or were present in infancy. These could lead to urinary tract complications presenting early in life and resulting in urinary tract infections. Treatments for hypotonic bladder may include increased fluid intake, techniques or drugs to trigger voiding, or surgery.
Ocular complications
Congenital mydriasis is a characteristic feature of SMDS detectable at birth as described previously.2 Eye complications have not been reported to lead to vision loss but patients may have varying degrees of poor vision. Complete eye examination by an ophthalmologist should be considered to assess vision and the need for glasses.2Funduscopic examination should be considered to assess arterial tortuosity and evolving complications.
Reproductive complications
Uncorrected cryptorchidism could lead to testicular atrophy requiring testosterone replacement therapy or azoospermia as observed in one patient and a previous report.27
Because uterine contraction is smooth muscle dependent, pregnant women may not be able to generate sufficient force to deliver vaginally. Given the high risk of aortic dissection or rupture, stroke, and other vascular complications associated with SMDS, discussion of these risks and evaluation by a cardiologist and high-risk obstetrician are strongly recommended before pregnancy. Genetic counseling should be part of the ongoing care of adolescent and young adult patients to discuss concerns about inheritance, recurrence risk, and reproductive options.
Limitations
Undiagnosed cases, including those with either milder or more severe sequelae who died before diagnosis, were not captured by this study. Incomplete or variable documentation of diagnoses and treatments in the medical records may have underestimated disease prevalence especially of acute or transient symptoms. Although the findings illustrate the range of complications associated with SMDS, future studies focused on defining specific complications, chronic issues, and quality of life are needed to fully understand SMDS and the needs of patients and caregivers.
Conclusions
ACTA2 Arg179 alterations cause a chronic condition presenting in infancy and requiring lifelong surveillance and treatment. Affected individuals are at risk of acute events due to aortic dissection or rupture, arterial thrombosis, ischemic stroke, and pulmonary complications. Multidisciplinary, coordinated care and surveillance, and timely management of symptoms are important to reduce the risk of SMDS complications. Future studies should address optimal imaging protocols to monitor the vascular disease and identify therapies to treat the vascular and pulmonary complications of SMDS.
References
Milewicz DM, Ostergaard JR, la-Kokko LM et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A 2010;152A:2437–2443.
Moller HU, Fledelius HC, Milewicz DM, Regalado ES, Ostergaard JR. Eye features in three Danish patients with multisystemic smooth muscle dysfunction syndrome. Br J Ophthalmol 2012;96:1227–1231.
Munot P, Saunders DE, Milewicz DM et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain. 2012;135:2506–2514.
Al-Mohaissen M, Allanson JE, O’Connor MD et al. Brachial artery occlusion in a young adult with an ACTA2 thoracic aortic aneurysm. Vasc Med. 2012;17:326–329.
Richer J, Milewicz DM, Gow R et al. R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet A 2012;158A:664–668.
Meuwissen ME, Lequin MH, Bindels-de HK et al. ACTA2 mutation with childhood cardiovascular, autonomic and brain anomalies and severe outcome. Am J Med Genet A 2013;161A:1376–1380.
Moosa AN, Traboulsi EI, Reid J, Prieto L, Moran R, Friedman NR. Neonatal stroke and progressive leukoencephalopathy in a child with an ACTA2 mutation. J Child Neurol 2013;28:531–534.
Amans MR, Stout C, Fox C et al. Cerebral arteriopathy associated with Arg179His ACTA2 mutation. J Neurointerv Surg 2014;6:e46.
Brodsky MC, Turan KE, Khanna CL, Patton A, Kirmani S. Congenital mydriasis and prune belly syndrome in a child with an ACTA2 mutation. J AAPOS. 2014;18:393–395.
Roulez FM, Faes F, Delbeke P et al. Congenital fixed dilated pupils due to ACTA2-multisystemic smooth muscle dysfunction syndrome. J Neuroophthalmol. 2014;34:137–143.
Yetman AT, Starr LJ, Bleyl SB, Meyers L, Delaney JW. Progressive aortic dilation associated with ACTA2 mutations presenting in infancy. Pediatrics. 2015;136:e262–e266.
Prabhu S, Fox S, Mattke A, Armes JE, Alphonso N. Extracorporeal life support in multisystem smooth muscle dysfunction syndrome. World J Pediatr Congenit Heart Surg 2017;8:750–753.
Logeswaran T, Friedburg C, Hofmann K et al. Two patients with the heterozygous R189H mutation in ACTA2 and Complex congenital heart defects expands the cardiac phenotype of multisystemic smooth muscle dysfunction syndrome. Am J Med Genet A 2017;173:959–965.
Ades LC, Davies R, Haan EA et al. Aortic dissection, patent ductus arteriosus, iris hypoplasia and brachytelephalangy in a male adolescent. Clin Dysmorphol. 1999;8:269–276.
Georgescu MM, Pinho MC, Richardson TE et al. The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathol Commun 2015;3:81.
Schneider DJ, Moore JW. Patent ductus arteriosus. Circulation. 2006;114:1873–1882.
Loeys BL, Dietz HC, Braverman AC et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476–485.
Lacro RV, Dietz HC, Sleeper LA et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014;371:2061–2071.
Regalado ES, Guo DC, Prakash S et al. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet 2015;8:457–464.
Anderson JL, Halperin JL, Albert NM et al. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA guideline recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127:1425–1443.
de Grazia J, Delgado I, Sanchez-Montanez A, Boronat S, Del CM, Vazquez E. Cerebral arteriopathy associated with heterozygous Arg179Cys mutation in the ACTA2 gene: report in 2 newborn siblings. Brain Dev. 2017;39:62–66.
Kikuchi A, Maeda M, Hanada R et al. Moyamoya syndrome following childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 2007;48:268–272.
Ullrich NJ, Robertson R, Kinnamon DD et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. 2007;68:932–938.
Arlachov Y, Ganatra RH. Sedation/anaesthesia in paediatric radiology. Br J Radiol 2012;85:e1018–e1031.
Roach ES, Golomb MR, Adams R et al. Management of stroke in infants and children—a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39:2644–2691.
Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst 2005;21:358–364.
Keir M, Al-Jundi W, Ouzounian M et al. Management of bilateral axillary aneurysms, threatened limb, and diffuse vasculopathy in a patient with ACTA2 mutation. Can J Cardiol 2017;33:554.
Acknowledgments
This research is supported by the National Institutes of Health (R01 HL109942), the John Ritter Foundation, the Genetic Aortic Disorders Association, and the Temerty Family Foundation. We are grateful to the patients and families who participated in this study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Regalado, E.S., Mellor-Crummey, L., De Backer, J. et al. Clinical history and management recommendations of the smooth muscle dysfunction syndrome due to ACTA2 arginine 179 alterations. Genet Med 20, 1206–1215 (2018). https://doi.org/10.1038/gim.2017.245
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.245
Keywords
This article is cited by
-
A yeast based assay establishes the pathogenicity of novel missense ACTA2 variants associated with aortic aneurysms
European Journal of Human Genetics (2024)
-
Nuclear smooth muscle α-actin participates in vascular smooth muscle cell differentiation
Nature Cardiovascular Research (2023)
-
Nonsurgical treatment of cerebral ischemia associated with ACTA2 cerebral arteriopathy: a case report and literature review
Child's Nervous System (2022)
-
Mapping a mammalian adult adrenal gland hierarchy across species by microwell-seq
Cell Regeneration (2020)
-
High-resolution iris and retinal imaging in multisystemic smooth muscle dysfunction syndrome due to a novel Asn117Lys substitution in ACTA2: a case report
BMC Ophthalmology (2020)
