
Abstract
Purpose
The objective of this study was to assess the ability of our laboratory’s exome-sequencing test to detect known and novel sequence variants and identify the critical factors influencing the interpretation of a clinical exome test.
Methods
We developed a two-tiered validation strategy: (i) a method-based approach that assessed the ability of our exome test to detect known variants using a reference HapMap sample, and (ii) an interpretation-based approach that assessed our relative ability to identify and interpret disease-causing variants, by analyzing and comparing the results of 19 randomly selected patients previously tested by external laboratories.
Results
We demonstrate that this approach is reproducible with >99% analytical sensitivity and specificity for single-nucleotide variants and indels <10 bp. Our findings were concordant with the reference laboratories in 84% of cases. A new molecular diagnosis was applied to three cases, including discovery of two novel candidate genes.
Conclusion
We provide an assessment of critical areas that influence interpretation of an exome test, including comprehensive phenotype capture, assessment of clinical overlap, availability of parental data, and the addressing of limitations in database updates. These results can be used to inform improvements in phenotype-driven interpretation of medical exomes in clinical and research settings.
Similar content being viewed by others

Introduction
Clinical laboratories are rapidly implementing next-generation sequencing (NGS)-based tests for the diagnosis of genetic disorders. While targeted, NGS-based gene panels are highly tuned to genes within a specific disease parameter, whole-exome sequencing (WES) tests assess a broad range of known and presumed phenotypes and genotypes. For example, there are clear clinical diagnostic guidelines and targeted gene tests available for Noonan syndrome.1, 2 In contrast, WES tests are applied without an a priori hypothesis to the majority of the coding regions in the human genome. Therefore, exome sequencing has become an important tool for diagnosing rare genetic conditions in patients with complex clinical presentations, or for whom previous testing has been either ambiguous or nondiagnostic.3 Even with expanded usage, exome-sequencing data continue to be challenging to analyze and interpret.4 Essential to the high-quality interpretation of an exome test is a carefully constructed analysis paradigm that can leverage phenotype data and family history in order to systematically characterize and interpret sequence variants in relation to a patient’s clinical indication for testing. Therefore, in addition to developing a strategy for variant identification and classification, analysis of exome data requires expertise in comprehensive review of thousands of genes, a challenging process to perform consistently across a wide variety of indications. Comprehensive validation strategies are therefore needed both to ensure the analytical validity of the test and to evaluate whether appropriate analysis strategies are being utilized, such that genomic data consistently receive valid interpretation.5, 6
Standards and guidelines are available to assist clinical laboratories with the validation of NGS-based tests.5, 6, 7, 8, 9 A number of laboratories have utilized these standards to establish analytical performance metrics for their clinical tests.10, 11, 12, 13 Validations for targeted disease panels typically include comparison of variant calls to a reference standard, and/or retesting biological specimens containing known pathogenic alterations.14, 15, 16 In contrast, validation of WES is typically performed by a methods-based validation approach, which includes comparison of variant calls to reference datasets of genome sequences.10 This method determines the exome test’s capacity for known variant detection (i.e., from DNA extraction to variant calling). It does not however, test critical-analysis processes that occur after variant calling, including the laboratory’s variant-analysis algorithms or the laboratory’s ability to leverage phenotype information for data interpretation.
Here, we describe a validation approach for WES that was designed not only to establish analytical performance criteria but also to rigorously test our interpretation process. As a first step, we assessed the ability of our sequencing and informatics process to detect known variants in a reference sample; we conducted this component of the validation by comparing the variants we detected to a benchmark set of high-confidence variant calls.17 We then tested our analysis and interpretation strategy by performing blinded exome analysis on 19 randomly selected probands who had previously undergone diagnostic exome sequencing by external reference laboratories, the reports of which served as our validation reference. Therefore, in order to evaluate concordance for “definitive” diagnoses, we compared our findings to those from the reference laboratories and explored the reasons underlying any discordances. Unique to this study is the emphasis on developing a systematic end-to-end validation approach for exome analysis from specimen preparation to interpretation. Our findings illustrate the complexities inherent in conducting comprehensive and reproducible phenotype-driven exome-interpretation strategies.
Materials and methods
Patients and blinded analysis strategy
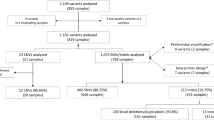
This study was performed on de-identified specimens and was exempted by the Children’s Hospital of Philadelphia Institutional Review Board. Of 70 patients, 19 were chosen by an honest broker for this study. The validation team was blinded to the reference laboratory results (Supplementary Materials and Methods online).
Extraction and sequencing
Libraries were prepared using the SureSelect Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA) and cluster generation and sequencing were performed using TruSeq Rapid SBS Kits–200 Cycle on a HiSeq 2500 (Illumina, San Diego, CA), following standard manufacturer’s guidelines.
Bioinformatics
Read alignment and variant calling were performed with an in-house bioinformatics pipeline incorporating NovoAlign (Novocraft, Selangor, Malaysia, http://www.novocraft.com/) for read alignment; Picard (Broad Institute, Cambridge, MA) for marking duplicates; and Genome Analysis Toolkit’s (GATK) (Broad Institute)18, 19 Best Practices for UnifiedGenotyper, with no parameter modifications (Supplementary Materials and Methods), for variant calling (reference sequence: hg19 Grch37) and variant filtering based on read depth (≥5 ×). Variant filtration was performed with Cartagenia Bench Lab (Agilent Technologies). Subsequently, a tiered approach, which incorporates phenotypic overlap, segregation information and variant information, was used to triage potential clinically significant variants in each patient’s dataset.
The medically relevant gene list is updated monthly by incorporating new gene-disease information from OMIM (Online Mendelian Inheritance in Man) and the Human Genome Mutation Database.20, 21
Analytical validation of single-nucleotide variant and indel detection
Using HapMap sample NA12878, we performed intra- and inter-run comparisons of single nucleotide variant (SNV) and indel calls in our data to determine repeatability and reproducibility. For intra-run comparisons, equimolar amounts of two libraries were prepared from NA12878, pooled and sequenced on the same HiSeq 2500 flowcell (runs 1a and 1b) and then repeated (runs 2a and 2b). Intra-run precision (repeatability) was evaluated by comparing our calls in each of the four runs to those in the National Institute of Standards and Technology–Genome in a Bottle Consortium (NIST-GIAB) data set (https://sites.stanford.edu/abms/giab).17 To evaluate inter-run precision (reproducibility), variants identified in NA12878 from four independent library preparation and sequencing runs were compared with the reference data set. Mean analytical sensitivity and specificity, and corresponding relative standard deviations were calculated for each intra- and inter-run comparison (Supplementary Materials and Methods and Supplementary Table S2).
Exome interpretation strategy
Variant filtration
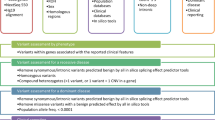
Variant annotation was performed using Cartagenia Bench Lab NGS, including gene, transcript, coding effect, predicted functional impact, minor allele frequencies, and the Human Genome Mutation Database. Variants (with a minor allele frequency of <3%) predicted to affect protein coding (missense, nonsense, frameshift, insertions, deletions, and splice site changes), along with any variants reported in the Human Genome Mutation Database, were analyzed. Where familial data were available, we prioritized variants matching an expected segregation pattern for genetic disease.
Exome data analysis
An interdisciplinary analysis team was established, consisting of molecular geneticists, physicians, fellows, genetic counselors, and laboratory scientists. Clinical presentation and family history were reviewed and leveraged to prioritize variants within genes with a high degree of clinical overlap. Analysis was restricted to variants within genes known or likely to be associated with human disease; review of variants in genes with unknown medical relevance was performed for negative cases. Each variant received a designation: (i) associated with phenotype, (ii) possibly associated with phenotype, (iii) not associated with phenotype, (iv) American College of Medical Genetics and Genomics secondary finding, or (v) candidate gene (not known to cause disease in humans). All variants other than (iii) were manually assessed and classified for pathogenicity, using an evidence-based review.22 Evidence and data collected during variant classification were gathered using Alamut Visual (Interactive Biosoftware, Rouen, France) and included information from various sources: population databases, computational assessments, ClinVar, the Human Genome Mutation Database, PubMed (https://www.ncbi.nlm.nih.gov/pubmed), and the University of California, Santa Cruz.20, 23, 24
Results
Validation of SNV and indel detection
We assessed our ability to detect known SNVs and indels 1–10 base pairs long in regions captured by the SureSelect Human All Exon V5 kit by performing intra- and inter-run comparisons using a reference sample (NA12878) to determine the analytical sensitivity and specificity for SNV and indel detection (Table 1). Mean sensitivity and specificity were greater than 99.4% and 99.9% respectively for SNV comparisons; indel comparisons had a mean sensitivity of >94% and mean specificity of 99.9%. For all six samples used in intra- and inter-run calculations, positive predictive values ranged from 99.8 to 99.9% for SNVs and 98.6 to 99.3% for indels. The false discovery rates were <0.2% for SNVs and <1.5% for indels (Supplementary Table S1).
We investigated discrepancies between the two datasets to determine whether the underlying causes of these discrepancies were due to known NGS errors. To do so, we performed an in-depth analysis of potential false negatives (46 SNVs and 60 indels) and false positives (19 SNVs and 6 indels) identified by our lab in run 1 (Table 2). BAM files were visually inspected using the Integrative Genomics Viewer (Broad Institute) for all potential false negatives and false positives. Insufficient coverage (<20 × depth) probably accounted for 78% of the false-negative SNV calls and 38% of the false-negative indel calls (Table 2). This study was restricted to indels ≤10 base pairs (bp), owing to the limited availability of indels >10 bp in the reference data set. However, we did observe that the sensitivity for indels 11–40 bp long dropped to 88.3% (data not shown). In addition, homopolymer stretches and highly repetitive regions probably accounted for 7% of SNV and 10% of indel false-negative calls. Allele bias and repetitive sequences appeared to account for 8% and 2% of false-negative indels, respectively, whereas we were unable to identify a possible underlying cause for 4% of the SNV and 12% of the indel false-negative calls. For the remaining 11% of SNVs and 30% of indels, a review of the BAM file alignment data supported the presence of a reference allele at those positions indicating possible errors in the reference data set used for the comparison. Sanger sequencing was performed on a select number of these variants, which further supported our call at these positions.
Subsequently, all putative false-positive calls were reviewed. Assessment of the BAM files suggested that 16% (3) of the potential false-positive SNVs were located in regions with insufficient coverage (<20 ×). The remaining 84% (16) SNVs and all six false-positive indels represent either potential true positives or are unique to the cell line (Table 2).
Validation of analysis strategy
Sequencing and analysis were performed on a cohort of 19 randomly selected pediatric patients for whom exome sequencing had been performed at external reference laboratories. Patients presented with a range of indications, including neurodevelopmental disorders (48%), multiple congenital anomalies (32%), metabolic disease (5%), skin (5%), immune (5%), and gastrointestinal disorders (5%). A single proband only was analyzed for 64% of the analyses. Of the remaining, 26% were trio-based (proband, mother, and father) and 10% were quad-based (proband, mother, father, and sibling) analyses (Supplementary Table S2).
In certain cases, more than one result was provided in patient reports, including multiple variants of uncertain significance. However, for the purpose of this study, the comparison of results was based on the presence or absence of a pathogenic/likely pathogenic variant with a strong correlation to the patient’s clinical indication. We also evaluated for concordance in cases where variants with a high indication of pathogenicity were reported in candidate genes by reference laboratories. Based on our experience, laboratories do not typically report candidate-gene variants as pathogenic, disease-causing mutations. Instead, they are included in a separate section of a report and do not constitute a positive diagnostic finding at the time of sign-out. Our reporting policy was consistent with this approach.
Overall, our findings were concordant with the reference laboratory for 84% (16/19) of patient reports (Table 3 and 4). Of the concordant findings, six individuals had a definitive molecular diagnosis (mutations in genes: GATA3, KANSL1, PEX1, SATB2, SMARCA2, and SYNGAP1), and ten were negative. For the three discordant cases, two had pathogenic variants, which were identified by both laboratories, but considered to be associated with the patient’s phenotype only by a reference laboratory (cases 3 and 16). The third discordant case did not have an initial diagnosis from the reference lab, but one was found during this validation study (case 11). Four of the nineteen cases had candidate-gene findings: two shared between the reference lab and this study (cases 7 and 8), and two unique to this study (cases 14 and 16). Overall, we identified new molecular findings in 16% of the cases (three patients), which were not reported by the reference laboratories at the time of this study.
Discordant cases
Case 3
The patient presented with bilateral cataracts and bilateral retinal detachment, severe bilateral sensorineural hearing loss, speech delay, panhypopituitarism, micropenis, undescended testes, and a family history of a maternal uncle with a small pituitary, hypothyroidism, and mild social and cognitive issues. A reference laboratory had previously reported a single heterozygous pathogenic variant as related to the individual’s phenotypic findings. This variant was in the B3GLCT gene, which is known to be associated with autosomal recessive Peters plus syndrome. This syndrome is characterized by anterior-chamber eye anomalies, growth deficiency with rhizomelic limb shortening, developmental delay, dysmorphic facial features, and cleft lip/palate.25 While our analysis did identify this pathogenic variant as “possibly associated with the ocular phenotype,” we did not consider this finding to have strong enough clinical overlap to account for the patient’s overall constellation of features. This assessment was corroborated by a clinical geneticist and a genetic counselor at CHOP. Both laboratories noted that a second pathogenic variant was not identified for this autosomal recessive condition. While the reference laboratory indicated that Peters plus syndrome was related to the indication for testing, our clinical-genetics team found the clinical correlation with Peters plus syndrome to be limited.
Case 16
The patient presented with epilepsy, hypotonia, sensorineural hearing loss, exotropia of the left eye, refractive amblyopia, reflux, chronic constipation, hyperextensibility, bruxism, and global developmental delay. A paternally inherited pathogenic variant was previously reported by an external reference laboratory, indicating a PRPH2-related disorder. PRPH2 is associated with several retinal dystrophies (retinitis pigmentosa, macular dystrophy, and cone rod dystrophy), which were not consistent with the patient’s clinical presentation at the time of testing. Moreover, the PRPH2 variant did not explain the patient’s neurodevelopmental phenotype (seizures, developmental delay, and hypotonia), an observation also noted in the report of the reference laboratory. A clinical geneticist from the study team was consulted and concurred with this correlation assessment. Therefore, while our analysis identified this variant, our laboratory classified it as having limited clinical correlation. Moreover, a novel candidate gene was identified during our analysis; this gene was later determined to be the molecular diagnosis for this patient (see below).
Case 11
The patient presented with Marfanoid habitus, intellectual disability, bilateral optic atrophy, and poor weight gain, with a decrease in adipose tissue and muscle mass. A clear molecular diagnosis was not identified by the reference laboratory. However, during our analysis a novel, de novo nonsense variant was found in the SOX5 gene (c.1021G>T; p.(G341*)). Upon further review of the literature, it was noted that intragenic deletions of SOX5 cause an intellectual-disability syndrome, phenotypically consistent with this patient.26 Therefore, SOX5 was established as the probable cause of the proband’s condition.27 This nonsense variant was also included on the reference laboratory’s report, but the gene was listed as not currently known to cause disease. At the time of analysis, SOX5 was not specifically annotated as a disease-causing gene in OMIM. Further, the reference laboratory had utilized only the proband’s DNA for exome sequencing, leaving the variant’s de novo inheritance unknown. These factors probably resulted in the discrepancies seen in individual 11’s exome interpretation.
Candidate genes
During the time of analysis, we encountered four cases with predicted loss-of-function variants in genes that had no previous association with human disease. Collaborative arrangements with other laboratories, literature searches, segregation data, and variant analysis led to the identification of novel candidate disease genes in these cases (Table 4).
Case 16
Our analysis and interpretation of the exome data in individual 16 indicated that the PRPH2 variant did not explain the clinical features (see above). Instead, we identified two rare compound heterozygous variants in trans in the gene SPATA5 (c.1343C>T; (p.Ser448Leu) and c.556C>T; p.(Arg186*)). Variants in this gene had not been reported to be associated with disease in humans at the time of our analysis. However, this gene possesses a high level of evolutionary conservation, and review of the Allen Brain Atlas website (http://www.brain-map.org/) indicated high levels of expression during brain development. Based on the lack of published evidence for human disease, we classified these as variants of uncertain significance in a novel candidate gene at the time of the validation study. Pathogenic variants in SPATA5 have since been recognized to be associated with seizures and intellectual disability.28, 29
Case 14
The patient presented with a history of nonimmune hydrops fetalis, developmental delay, proportionate short stature, lumbar vertebral anomalies, mild idiopathic pulmonary hypertension, long-segment tracheal stenosis, coloboma, microphthalmia, visual impairment, hydrometrocolpos, pelviectasis, hydronephrosis, persistent urogenital sinus and clitoromegaly, external ear malformation with conductive hearing loss and ear pits, and mild hypotonia. No disease-causing variant was identified by the reference laboratory. However, during our analysis, a novel, de novo nonsense variant was found in the gene KAT6A (c.3116_3117 delCT; (p.Ser1039∗)). This gene possesses a high level of evolutionary conservation, and has an animal model which demonstrates craniofacial and cardiac anomalies. Through the use of a social-media networking strategy we were able to identify a similar patient with a de novo KAT6A mutation.30 While we called this finding a possibly causal variant in a candidate gene at the time of the validation study, our genetic testing facility, along with several others, was later able to publish a cohort of patients with KAT6A mutations and a shared syndromic phenotype.30
The last two candidate gene findings were reported by both the reference laboratory and this study. We identified the gene ATAD1 as a candidate gene in case 7 through segregation analysis and review of knockout mouse models.31 For case 8, we reported WDR26 as a candidate gene after review of segregation and comparison with deletion syndrome reports.32
Discussion
Using NA12878 (curated by NIST-GIAB)15, 17 as a comparison to calculate sensitivity, specificity, repeatability, reproducibility, positive predictive value, and false discovery rate, we were able to demonstrate the consistently high quality of our exome data (Table 1). These results demonstrate that the process spanning procedures from DNA extraction to variant calling is robust and reliable. Our interpretation strategy was tested by comparing results from our complete analysis of 19 exomes to prior results obtained by other reference laboratories, and 84% concordance was found ( Table 3 ). In three patients (11, 14, and 16) new molecular diagnoses were reached (two involving novel, candidate genes), and this further highlights the utility of continuing to reanalyze exomes over a period of time. These findings further highlight the need for efficient and cost-effective exome reanalysis strategies, as novel gene-disease associations are identified frequently and existing associations are updated continuously.5, 33, 34
Overall, we determined that our reported results were consistent with those of other laboratories (16/19; 84% concordance) and that the sources of the discrepancies were not unexpected, given the complex nature of the analytic process and the reliance on phenotype information. For instance, access to the experienced clinical geneticists and to detailed phenotypic information as a resource for analysis contributed to the discrepancies seen in cases 3, 11, and 16. Based on these results, we conclude that exome interpretation is influenced by several critical factors, including the availability of detailed phenotypic data, availability of parental exome data, and the ability to integrate complex phenotype information with evolving gene-disease information.
We acknowledge that laboratories may differ in whether they report variants in genes for which there is limited or no evidence suggesting an association with human disease. In addition, the laboratory must decide what to report, based on overlap with an individual’s clinical indication. The definition of clinical overlap also differs between laboratories. One laboratory may report a finding with minimal phenotypic overlap, whereas another laboratory may report only findings that match most of the clinical indication provided. The variety of approaches to analyses and the subjectivity in reporting decisions could lead to a patient’s receiving different results depending on the laboratory, as evidenced by this validation study. Thus, educating ordering providers is important, as they may not be aware of pertinent differences among laboratory testing ideologies and the importance of critical phenotype data. An open discussion among members of the exome-sequencing community about the benefits and limitations of these strategies would be beneficial.
Exome sequencing can yield a large number of variants for analysis, a rate-limiting step for many clinical laboratories. Rapid and accurate identification of variants in genes that are most relevant to the phenotype of the patient is crucial, influencing the sensitivity and turnaround time of this test. Access to the patient’s clinical presentation, and past medical and family history is critical, since this information can be leveraged to mine gene-disease databases to prioritize variants within genes with a high degree of clinical overlap. The success of a phenotype-driven analysis relies on several critical assumptions, (i) clinical information and past medical history is accessible to the laboratory team, (ii) the laboratory has expertise in assessment of diverse and complex clinical presentations, and (iii) the laboratory has access to reliable and sensitive approaches to prioritizing clinically relevant variants. However, there are several barriers to this process.
Access to electronic health records is not typically available for reference laboratories. To address this limitation, medical records are frequently provided to the testing laboratory by the physician. However, these records may be incomplete and the manual chart-review process in itself can be labor-intensive and frequently necessitates extensive clinical expertise, covering a wide range of clinical indications, not typically available in a diagnostic laboratory.
Data analysis can be performed using both manual and informatics procedures to leverage phenotype for prioritizing analysis of clinically relevant genes. One approach that is likely to be labor-intensive is for a laboratory to generate a customized gene list specific to the patient’s clinical indication by using specific terms to interrogate various gene-disease databases (OMIM). However, owing to lack of usage of standardized phenotype terminology, and limitations in updating new information in various databases, this process can be error-prone and labor-intensive, and is therefore not scalable. An alternative to the manual approach is to utilize informatics approaches by leveraging standardized phenotype-ontology terms to prioritize genes based on the patient’s phenotype and sequence information.35 In this case, the laboratory needs substantial clinical expertise to translate the terms used to describe the patient’s key clinical features into standardized ontology terms.
In order to facilitate scalable and sensitive interpretation of data, the exome-testing approach was modified and revalidated to include several enhancements. When practical, we encourage clinicians to provide biological parental samples, in order to facilitate sensitive identification of potentially pathogenic variants by use of the trio exome-analysis method. We have also developed custom algorithms to leverage standard Human Phenotype Ontology terms,36 variant type, and segregation data by highlighting potentially significant variants in genes that are relevant to the patient’s phenotype (unpublished material). We have established a collaborative arrangement with the clinical geneticists and genetic counselors within the Roberts Individualized Medical Genetics Center at CHOP. This group plays an important role in the analysis of exome data through an exhaustive review of the patient’s chart and data analysis in the form of gene-disease correlation assessment. The center performs a systematic review of all systems, including capture of the patient’s clinical presentation using Human Phenotype Ontology32 terms, assessment of family history, and analysis of prior testing records. We believe that this arrangement, in combination with the algorithmic approach, leverages the combined expertise of laboratory, informatics, and clinical teams to provide enhanced clinical interpretation of the exome data.
While differences in methodology for collection and utilization of phenotypic data may lead to variability in reporting, the diagnostic rate for WES is high, in comparison to other technologies (reaching 25–30% diagnostic yield) and the clinical utility of WES has been described for a variety of testing conditions.37, 38, 39 In addition, since exome sequencing provides genome-wide data, reanalysis in the years after initial testing can lead to more diagnoses.39 Based on our validation study, concordance was nearly complete between testing laboratories for cases where a full molecular diagnosis was identified (cases 2, 4, 6, 12, 17, and 18). The cases in which a new molecular diagnosis was found during validation included instances where new data were included, either from sequencing family members or from newly emerging disease information.
In conclusion, we report a two-step validation approach to rigorously test the identification, analysis, and interpretation of variants by exome sequencing, with the first step a technical review, and the second step an analysis and reporting review. Our experience highlights not only the diagnostic utility of WES, especially with regard to rare and novel genetic disorders, but also the potential sources of variability in variants selected for exome-sequencing reports. It also exposes important considerations that influence interpretation for laboratories currently performing clinical WES.
References
van der Burgt I . Noonan syndrome. Orphanet J Rare Dis 2007;2:4.
Aoki Y, Niihori T, Inoue S, Matsubara Y . Recent advances in RASopathies. J Hum Genet 2016;61:33–39.
Valencia CA, Husami A, Holle J et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr 2015;3:67.
Seaby EG, Pengelly RJ, Ennis S . Exome sequencing explained: a practical guide to its clinical application. Brief Funct Genomics 2015;15:374–84.
Rehm HL, Bale SJ, Bayrak-Toydemir P et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013;15:733–747.
Gargis AS, Kalman L, Berry MW et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol 2012;30:1033–1036.
Schrijver I, Aziz N, Farkas DH et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J Mol Diagn 2012;14:525–540.
MacArthur DG, Manolio TA, Dimmock DP et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476.
Aziz N, Zhao Q, Bry L et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med 2015;139:481–493.
Linderman MD, Brandt T, Edelmann L et al. Analytical validation of whole exome and whole genome sequencing for clinical applications. BMC Med Genomics 2014;7:20.
Lepri FR, Scavelli R, Digilio MC et al. Diagnosis of Noonan syndrome and related disorders using target next generation sequencing. BMC Med Genet 2014;15:14.
Simen BB, Yin L, Goswami CP et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med 2015;139:508–517.
Grosu DS, Hague L, Chelliserry M et al. Clinical investigational studies for validation of a next-generation sequencing in vitro diagnostic device for cystic fibrosis testing. Expert Rev Mol Diagn 2014;14:605–622.
Baudhuin LM, Lagerstedt SA, Klee EW, Fadra N, Oglesbee D, Ferber MJ . Confirming variants in next-generation sequencing panel testing by Sanger sequencing. J Mol Diagn 2015;17:456–461.
Vasli N, Bohm J, Le Gras S et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012;124:273–283.
Wang J, Zhang VW, Feng Y et al. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest Ophthalmol Vis Sci 2014;55:6213–6223.
Zook JM, Chapman B, Wang J et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32:246–251.
McKenna A, Hanna M, Banks E et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Van der Auwera GA, Carneiro MO, Hartl C et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43 (11 10 11-33).
Stenson PD, Ball EV, Mort M et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581.
Online Mendelian Inheritance in Man OM-NIoGM. Accessed 2013-present. https://omim.org/.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Speir ML, Zweig AS, Rosenbloom KR et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res 2016;44(D1):D717–725.
Landrum MJ, Lee JM, Benson M et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44(D1):D862–868.
Lesnik Oberstein SAJ, van Belzen M, Hennekam R. Peters Plus Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. University of Washington: Seattle, WA, 1993.
Schanze I, Schanze D, Bacino CA, Douzgou S, Kerr B, Zenker M . Haploinsufficiency of SOX5, a member of the SOX (SRY-related HMG-box) family of transcription factors is a cause of intellectual disability. Eur J Med Genet 2013;56:108–113.
Nesbitt A, Bhoj EJ, McDonald Gibson K et al. Exome sequencing expands the mechanism of SOX5-associated intellectual disability: a case presentation with review of sox-related disorders. Am J Med Genet A 2015;167A:2548–2554.
Tanaka AJ, Cho MT, Millan F et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 2015;97:457–464.
Buchert R, Nesbitt AI, Tawamie H et al. SPATA5 mutations cause a distinct autosomal recessive phenotype of intellectual disability, hypotonia and hearing loss. Orphanet J Rare Dis 2016;11:130.
Tham E, Lindstrand A, Santani A et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am J Hum Genet 2015;96:507–513.
Ahrens-Nicklas RC, Umanah GK, Sondheimer N et al. Precision therapy for a new disorder of AMPA receptor recycling due to mutations in ATAD1. Neurol Genet 2017;3:e130.
Shaffer LG, Theisen A, Bejjani BA et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616.
Christensen KD, Dukhovny D, Siebert U, Green RC . Assessing the costs and cost-effectiveness of genomic sequencing. J Pers Med 2015;5:470–486.
Wenger AM, Guturu H, Bernstein JA, Bejerano G . Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2016;19:209–214.
Robinson PN, Kohler S, Oellrich A et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014;24:340–348.
Robinson PN, Kohler S, Bauer S, Seelow D, Horn D, Mundlos S . The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet 2008;83:610–615.
Stark Z, Schofield D, Alam K et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867–874.
Stark Z, Tan TY, Chong B et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016;18:1090–1096.
Vissers LE, van Nimwegen KJ, Schieving JH et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med 2017;19:1055–1063.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AS work for a fee-for-service laboratory to perform clinical genetic testing. AS serves on advisory capacity or in other capacities for Veritas, CambridgeHealth Tech, Arcadia University and receives licensing and royalties from Agilent.
Additional information
The first two authors contributed equally to this work.
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
About this article

Cite this article
Gibson, K., Nesbitt, A., Cao, K. et al. Novel findings with reassessment of exome data: implications for validation testing and interpretation of genomic data. Genet Med 20, 329–336 (2018). https://doi.org/10.1038/gim.2017.153
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.153
Keywords
This article is cited by
-
Interplay between probe design and test performance: overlap between genomic regions of interest, capture regions and high quality reference calls influence performance of WES-based assays
Human Genetics (2021)
-
A highly sensitive and specific workflow for detecting rare copy-number variants from exome sequencing data
Genome Medicine (2020)
-
A flexible computational pipeline for research analyses of unsolved clinical exome cases
npj Genomic Medicine (2020)
-
Increased diagnostic yield by reanalysis of data from a hearing loss gene panel
BMC Medical Genomics (2019)
-
Rapid and accurate interpretation of clinical exomes using Phenoxome: a computational phenotype-driven approach
European Journal of Human Genetics (2019)
