
Abstract
Purpose
Genetic testing is an integral diagnostic component of pediatric medicine. Standard of care is often a time-consuming stepwise approach involving chromosomal microarray analysis and targeted gene sequencing panels, which can be costly and inconclusive. Whole-genome sequencing (WGS) provides a comprehensive testing platform that has the potential to streamline genetic assessments, but there are limited comparative data to guide its clinical use.
Methods
We prospectively recruited 103 patients from pediatric non-genetic subspecialty clinics, each with a clinical phenotype suggestive of an underlying genetic disorder, and compared the diagnostic yield and coverage of WGS with those of conventional genetic testing.
Results
WGS identified diagnostic variants in 41% of individuals, representing a significant increase over conventional testing results (24%; P = 0.01). Genes clinically sequenced in the cohort (n = 1,226) were well covered by WGS, with a median exonic coverage of 40 × ±8 × (mean ±SD). All the molecular diagnoses made by conventional methods were captured by WGS. The 18 new diagnoses made with WGS included structural and non-exonic sequence variants not detectable with whole-exome sequencing, and confirmed recent disease associations with the genes PIGG, RNU4ATAC, TRIO, and UNC13A.
Conclusion
WGS as a primary clinical test provided a higher diagnostic yield than conventional genetic testing in a clinically heterogeneous cohort.
Similar content being viewed by others

Introduction
Over the last decade, advances in high-throughput sequencing technologies have had a considerable impact on clinical genetic testing.1, 2 There has been increased recognition and expansion of the role that genetic testing plays in pediatric medicine, and tests are commonly ordered by non-geneticist subspecialists. Contemporary testing for genetically heterogeneous phenotypes often consists of a combination of chromosomal microarray analysis (CMA) to detect copy number variation (CNV) and targeted next-generation sequencing (NGS) gene panels to detect single-nucleotide variants (SNVs) and small insertions and deletions (indels). These conventional stepwise strategies can be costly and time-consuming, and yet for most disease cohorts they yield a molecular diagnosis in only a small fraction of patients. Clinical hypothesis-driven approaches can lead physicians to restrict their focus to a specific organ system or phenotype component, and to limit testing to a priori constructed gene panels that may or may not reflect the full differential diagnosis. Genome-wide analysis by clinical whole-exome sequencing (WES) has dramatically increased the diagnostic yield in individuals with suspected genetic disorders.3, 4, 5 However, prospective studies are few,6 and WES can miss major types and regions of disease-causing genomic variation (e.g., indels, structural variants, intronic SNVs).
Unlike WES, whole-genome sequencing (WGS) offers the potential of a single test that captures nearly all genomic variation in an unbiased manner. There is emerging evidence for its utility in clinical diagnosis and in gene discovery.1, 7, 8, 9, 10 Much speculation centers on the perceived advantages and limitations of genome-wide testing relative to targeted testing. Healthy skepticism among clinicians about the feasibility of testing with WGS is fueled in part by unanswered questions regarding the scalability of clinical analytic pipelines, the prospective diagnostic yield, and test sensitivity compared with that of standard-of-care testing. The Genome Clinic at The Hospital for Sick Children (Toronto, ON, Canada) is a longitudinal multifaceted research project designed to integrate WGS into mainstream pediatrics.11 In our initial study, we found evidence for the diagnostic superiority of WGS to conventional genetic testing, ordered by clinical geneticists, in a cohort of patients who met the criteria for CMA.1 Here, we report our prospective comparison of WGS and NGS gene panels and other routine testing in 103 new patients with diverse phenotypes, drawn from a range of pediatric non-genetics subspecialty clinics.
Materials and methods
Participant recruitment and inclusion/exclusion criteria
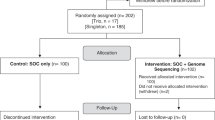
We recruited unrelated patients ≤18 years old from pediatric subspecialty clinics at The Hospital for Sick Children (Toronto, ON, Canada) over a 2-year period (April 2013 to June 2015). As a complement to our previous study,1 a roughly equal number of participants were purposefully recruited from outside the Clinical Genetics clinic. Patients without a molecular genetic diagnosis were eligible to participate in this study if they met the following criteria:
-
1
They were being followed in a subspecialty outpatient clinic at The Hospital for Sick Children
-
2
Their disease was well characterized clinically and was known to be genetically heterogeneous
-
3
The standard of care at the time of recruitment was to request genetic testing to assist in diagnosis and disease management
-
4
Clinical genetic testing was to involve examination of multiple genes
-
5
The existing multigene testing had incomplete sensitivity
We also required that both parents be available for testing and, because of the complexity of the consenting process, that they be fluent in English. Patients and families were offered the option of learning about secondary variants related to adult-onset disease risk.12 The study was approved by the Research Ethics Board at The Hospital for Sick Children. Informed written consent was obtained for each participant. Of the 113 individuals who were initially consented into the study, four (cases 26, 62, 73, and 85) withdrew prior to WGS, five (cases 23, 24, 31, 35, and 61) did not meet the eligibility criteria, and one (case 15) was excluded because of poor DNA quality.
Phenotyping and conventional diagnostic testing
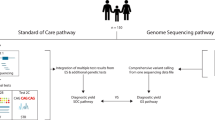
Phenotype data were extracted from the electronic medical record and entered into PhenoTips (http://www.phenotips.org) (Gene42, Toronto, ON, Canada), an open-source software program for collecting and analyzing phenotypic information for patients with genetic disorders.13 Phenotypic information is represented in PhenoTips using the Human Phenotype Ontology (HPO). Data regarding conventional molecular genetic testing ordered by attending clinicians (and the associated cost in US dollars at that time, when available) were also extracted from the electronic medical record. By design, all individuals had had targeted gene sequencing. A significant minority (43%) were also tested with CMA. Supportive investigations such as chemistry tests (blood and urine), enzymatic studies, muscle biopsies, and medical imaging were noted but not considered in cost analyses.
WGS
WGS of index participants was performed with the Illumina (San Diego, CA) HiSeq X system at The Centre for Applied Genomics in Toronto, Ontario, Canada from DNA extracted from whole blood. DNA was quantified using the Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA) High Sensitivity Assay, and sample purity was checked using the Nanodrop (Thermo Fisher Scientific, Waltham, MA) OD 260/280 ratio. Following the manufacturer’s recommended protocol, 100 ng of DNA were used as input material for library preparation using the Illumina TruSeq Nano DNA Library Prep Kit. In brief, DNA was fragmented to an average of 350 base pairs by sonication on a Covaris (Woburn, MA) LE220 instrument. Fragmented DNA was end-repaired and A-tailed and indexed TruSeq Illumina adapters added by ligation before library amplification. Libraries were assessed using Bioanalyzer DNA High Sensitivity chips (Agilent Technologies, Santa Clara, CA) and quantified by quantitative polymerase chain reaction using the Kapa Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems, Roche, Basel, Switzerland). Validated libraries were pooled in equimolar quantities and paired-end sequenced on an Illumina HiSeq X platform, following Illumina’s recommended protocol, to generate paired-end reads of 150 bases in length.
Variant calling and annotation
Base calling and data analysis were performed using BCL2FASTQ, and data were analyzed using Illumina HiSeq Analysis Software (HAS; version 2-2.5.55.1311). Reads were mapped to the hg19 reference sequence using Isaac Genome Alignment Software (SAAC00776.15.01.27) (Illumina) and SNVs and small indel variants were called using Starling (Isaac Variant Caller; version 2.1.4.2).14 WGS data will be deposited in the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/). Resulting variant calls were annotated using a custom pipeline1 developed at The Centre for Applied Genomics, based on ANNOVAR.15 Mitochondrial variants were converted to NC_012920 coordinates with a custom script and then annotated using MitImpact1916 (version 2.4, http://mitimpact.css-mendel.it/) to identify known pathogenic variants. CNVs were called, using the read-depth method, by the programs ERDS (Estimation by Read Depth with Single-Nucleotide Variants)17 and CNVnator,18 using a window size of 500 base pairs. CNV size cutoffs were 1 kb for losses and 2 kb for gains. High-quality CNVs were defined as those detected by ERDS that were also detected by CNVnator with greater than 50% overlap.
Clinical interpretation and confirmation of variants
As in our previous study,1 molecular and clinical geneticists examined variant files and prioritized clinically relevant nuclear DNA variants in index participants using the following parameters: (i) sequence quality, (ii) allele frequency, (iii) conservation and predicted impact on coding and noncoding sequence, (iv) presence in ClinVar19 or the Human Gene Mutation Database (HGMD),20 (v) zygosity and genetic mode of inheritance, and (vi) relevance to the reported clinical features. Percent heteroplasmy of known pathogenic mitochondrial DNA variants was estimated using read counts. WGS was performed under a research protocol and not as a validated clinical test. However, candidate pathogenic variants deemed relevant to the primary phenotype according to established laboratory reporting criteria21 were discussed with the referring clinician and designated as diagnostic by consensus. Some diagnostic variants, including all the mitochondrial DNA variants, were confirmed by conventional genetic tests ordered by the clinicians. Diagnostic variants not found through conventional testing were confirmed by Sanger sequencing or quantitative polymerase chain reaction in a laboratory with Clinical Laboratory Improvement Amendments/College of American Pathologists certification, and a clinical report was generated. Inheritance of variants was determined via targeted analysis of parental DNA samples. In total, six candidate variants were deemed non-diagnostic on the basis of segregation testing.
Statistical methods
We compared the diagnostic yield of WGS to conventional genetic testing using a chi-square proportion test. Differences between subgroups with respect to clinical and demographic characteristics were assessed using Fisher’s exact test or a chi-square test for categorical variables and the Mann–Whitney U test for continuous variables. All tests were two-tailed, with statistical significance defined as P < 0.05.
Results
Cohort characteristics
In total, 103 individuals were included in the study (Table 1). Basic demographic characteristics were representative of the clinic populations from which participants were recruited. Similar to those in the previous study cohort recruited from our hospital,1 nine (8.7%) individuals were offspring of consanguineous unions. Referrals were received from 10 clinics, and the majority (86%) of individuals had been seen in more than one subspecialty clinic (Table 1). By design, no patients were recruited from the Clinical Genetics clinic, but 10.7% were seen there on at least one occasion. Patients displayed a wide array of symptoms, described by 647 unique HPO terms across the cohort. The five most commonly represented HPO categories were neurological (n = 70; 68.0%); musculoskeletal (n = 54; 52.4%); eye defect (n = 52; 50.5%); behavior, cognition, and development (n = 50; 48.5%); and “other” (n = 49; 47.6%). The median number of HPO categories per individual was 5 (Table 1), and each HPO category was represented in >20% of participants.
Description of conventional genetic testing
The median number of conventional genetic tests was 3 and the median number of nuclear genes sequenced was 19 (Table 1). These tests were primarily NGS targeted gene panels and resulted in the sequencing of 1,226 different genes. However, the single most common genetic test was CMA (n = 44 participants), which revealed variants of unknown significance in six participants and no diagnostic variants (Supplementary Table S1 online). Costs were known for 136 (76%) of the unique tests. In 100 of 103 participants with costs available for at least one test, the median cost per individual for this clinical genetic testing was US$5,173 (Table 1).
Genetic tests beyond the scope of WGS, and thus not considered in the above analyses, included karyotype (n = 19), polymerase chain reaction for triplet repeat expansion (n = 14), multiplex ligation-dependent probe amplification for imprinting diseases (n = 12), chromosome breakage studies (n = 3), X chromosome inactivation studies (n = 1), and fluorescence in situ hybridization on fibroblasts for mosaic trisomy 8 (n = 1). In addition, nine participants had clinical WES. None of these tests resulted in a diagnosis.
Yield of conventional genetic testing
Twenty-five study participants (24%) obtained a molecular diagnosis via conventional genetic testing (Supplementary Table S2). This included three participants who received a partial diagnosis (i.e., the clinician may continue to pursue genetic testing to explain other aspects of their phenotype). The disease inheritance pattern was: autosomal recessive (n = 14), autosomal dominant (n = 6), mitochondrial (n = 3), and X-linked (n = 2). There was heterogeneity across referral clinics in the diagnostic yield of standard-of-care genetic investigations; for example, 11 were diagnosed from the Ophthalmology clinic22 (yield of 46%) and none were diagnosed from the Joint laxity/hypermobility clinic. The median number of genetic tests, median number of genes sequenced, and median total cost of testing per person in the n = 25 who received a diagnosis were comparable to those for the remaining cohort (data not shown).
Summary statistics and WGS coverage
On average across the cohort, the mean and median depth coverage of WGS was 37 × (Supplementary Table S3). Genome-wide coverage at 10 × and 20 × was 98% and 93%, respectively (Supplementary Table S3). WGS generated a mean of 3.9 million high-quality SNVs and indels (21,705 coding) per individual, including 255,388 (1,311 coding) variants that were rare (<5% population frequency in publicly available databases1) (Supplementary Table S4). The average number of high-quality rare CNVs per sample was 17 (5 coding) (Supplementary Table S5).
We also examined the WGS coverage of the 16,810 exons of the 1,226 total genes sequenced by conventional methods in the study participants. Every individual had a median exonic coverage (MEC) ≥18 ×, and the mean MEC across the cohort was 40 × (Supplementary Figure S1). With respect to the standard deviation in MEC within an individual, the median across the cohort was 8.3 ×, consistent with relatively uniform coverage (Supplementary Figure S1). Overall, the vast majority of the 16,810 exons had ≥10 × coverage (Supplementary Figure S2), suggesting that WGS has acceptable sensitivity. The 15 exons with a mean coverage of less than 10 × across the cohort (indicating a potential for false negatives) are listed in Supplementary Table S6.
Diagnostic yield of WGS
WGS identified diagnostic variants in 42 study participants (41%), representing a significant increase over conventional testing (P = 0.01). Seventeen participants with diagnoses made only via WGS are described in Table 2. Case 36 had pathogenic variants in two different genes that contributed to her phenotype. The disease inheritance pattern of the 18 total genes was: autosomal recessive (n = 8), autosomal dominant (n = 5), and X-linked (n = 5). In addition, two participants had reportable variants that may explain a single (minor) aspect of a multisystem phenotype, and two had strong candidate variants that will be pursued on a research basis (data not shown). All diagnostic variants discovered with conventional testing (Supplementary Table S2), including the mitochondrial DNA variants, and all reportable CNVs on CMA (Supplementary Table S1), were also identified with WGS. Orthogonal confirmation of these WGS findings was thus obtained from the conventional tests that resulted in the clinical diagnoses. As expected,5 participants from consanguineous families were significantly overrepresented in the cohort with molecular diagnoses (Table 1). Seven of these nine children (78%) received a diagnosis, and the remaining two were homozygous for variants of uncertain significance (data not shown). The distribution of referral clinics was also significantly different between the diagnosed and undiagnosed groups (Table 1), in a pattern broadly consistent with results from a previous study.5
Case-level analysis of WGS diagnoses
The variants in Table 2 are mostly exonic SNVs in genes that were never tested via conventional testing, either because the discovery of the disease association occurred shortly before or after the NGS gene panel testing, or because the testing was not broad enough. For example, case 64 had extensive biochemical and genetic testing for mitochondrial disorders on the basis of a muscle biopsy early in life reporting an isolated complex II deficiency. However, he was ultimately found to have a diagnostic variant in PIGG,23 an endoplasmic reticulum gene that would probably not have been considered for clinical testing.
The results of this study provide valuable validation for several emerging disease genes, including PIGG,23 RNU4ATAC,24 TRIO,25 and UNC13A.26 In addition, the majority of variants in Table 2 are novel. In two participants (cases 3 and 45), pathogenic deep intronic variants with prior published experimental evidence were identified with WGS and missed by conventional sequencing (Table 2). Case 5 was compound heterozygous for SNVs in the small nuclear RNA gene RNU4ATAC, which is not targeted on most commercially available exome capture kits.24 Structural variants were a minor contributor: case 11 was compound heterozygous for a SNV and a small (4.5 kb) CNV disrupting SLC25A19 (Table 2). Of note, both this CNV and the one in case 63 (Supplementary Table S2) are of a size not routinely detectable with genome-wide CMA and would require targeted testing. Genetic counseling was provided to all individuals and their families, and potential treatment implications were reviewed with the referring physician.
Comparing WGS and WES
To compare the diagnostic yield of WGS with WES, the first 70 participants who provided DNA samples (68.0% of the final study cohort) also underwent research-based WES to a mean depth of coverage >100 × using the Ion Proton system, following exonic amplification with the Ion AmpliSeq Exome Kit (Life Technologies). This included 35 of the 42 participants who ultimately received a genetic diagnosis during the study period (Table 2 and Supplementary Table S2). WES methods were as described previously.27 Not only did WGS detect all diagnostic variants found by WES, but in 9 (25.7%) of the 35 participants WGS revealed diagnostic variants not apparent in the WES data: cases 3, 5, 11, and 45 (Table 2), and cases 4, 10, 29, 49, 59, and 63 (Supplementary Table S2). These variants included deep intronic SNVs (cases 3 and 45), small CNVs (cases 11 and 63), SNVs in a noncoding RNA (case 5), mitochondrial DNA variants (cases 4 and 49), and exonic SNVs in regions with poor coverage on WES (cases 10 and 29). In the latter two instances, there were technical issues that may not be applicable to other exome enrichment methodologies (data not shown).
Discussion
The results provide evidence for incorporating WGS in the clinical workup of children with suspected genetic disorders. Moreover, these prospective data support a testing strategy that involves early utilization of genome-wide analyses. Using an established clinical analytic pipeline,1 WGS increased overall genetic diagnostic yield while also detecting all disease-causing variants identified by conventional testing strategies. For many patients, the diagnostic odyssey, which consists of both quantifiable (e.g., time; costs of genetic and extra-genetic investigations) and unquantifiable burdens, could be shortened with the early use of WGS.
The application of WGS in clinical practice
As noted above, many of the new diagnoses attributable to WGS were the result of specific technological advantages over conventional testing. An additional advantage is the opportunity for periodic systematic re-annotation of genome-wide variants.5, 6, 28 For some of the undiagnosed individuals in this cohort, we anticipate that causal mutation(s) will be identified within the next 2–3 years without the need to perform additional genetic testing. The alternative—performing new single-gene testing or repeating gene panel testing with each discovery of a new candidate gene—is inefficient, time-consuming, and costly. Consistent with previous reports,1, 3, 4, 5 the one individual with two diagnostic variants (Table 2) and the five with partial diagnoses suggest that 4–5% of this cohort may have more than one explanatory genetic mutation. WGS data can also be mined for medically significant pharmacogenomic variants and predictive secondary variants. If symptoms suggestive of another genetic condition emerge later in life, preexisting WGS data could be interrogated in place of new testing.
Lessons learned from the clinical application of WES may be generalizable to WGS. The initial pattern of practice was often to reserve WES for patients who remained undiagnosed after considerable targeted genetic testing. In our study, clinicians ordered clinical WES infrequently (for 9 of 103 participants; 8.7%) and late in the diagnostic workup. Emerging prospective data suggest that the early use of WES in diagnostic evaluations can result in cost savings and improved diagnostic yields, compared to conventional genetic and non-genetic investigations.6, 29 Our data suggest WGS may offer similar benefits to WES but with even higher diagnostic yields. Time to diagnosis after initiating genetic testing was not recorded in this study. However, even in individuals ultimately diagnosed by conventional testing we suspect that WGS would have on average arrived at a diagnosis first (if initiated at the same time), because of the delays inherent in sequential testing strategies.
While WGS is frequently described as hypothesis-free, knowledge of both the complete genotype and the phenotype can be used to iteratively generate, test, and refine hypotheses that lead to a diagnosis. This is one of the clear advantages of WGS over conventional genetic testing, in which the clinical phenotype drives the generation of hypotheses that determine which genes are examined. WGS is not a diagnostic panacea, however. Accurate and comprehensive phenotyping is critical, as the ability to generate a differential diagnosis facilitates efficient interpretation of WGS data. Unidirectional clinical hypothesis-driven testing can have many advantages—e.g., fewer variants of uncertain significance and (usually) lower cost—in settings where the pretest probability of a specific mono- or oligo-genetic disorder is very high. In addition, certain molecular mechanisms (e.g., triplet repeat expansions) test the limits of contemporary WGS. A better understanding of the clinical utility, value for money, and associated ethical and societal implications of WGS will also need to be answered prior to its widespread clinical use.
WGS is diagnostically superior to WES
WES has emerged as a powerful genetic diagnostic tool. However, one concern about clinical WES, in comparison to targeted testing, is the potential for inadequate coverage of some exons of essential candidate genes for the presenting phenotype. This is less of a concern with WGS, because at typical sequencing depth it offers improved uniformity of coverage of exonic regions compared to WES.30 Our data demonstrating excellent coverage of targeted exons suggest WGS would have sufficient coverage of tested exons compared to targeted NGS gene panels. The very small fraction of exons not well covered in a typical WGS experiment can be backfilled via Sanger sequencing on an as-needed basis.
The diagnostic yield of WGS in this study was the result of its ability not only to identify those diagnostic variants detectable by WES but also to detect diagnostic variants beyond the scope of WES. These included intronic SNVs, SNVs in noncoding RNA, small CNVs, and mitochondrial DNA mutations, as well as exonic SNVs missed as a consequence of WES coverage and enrichment methods. Over time, the ability to interpret deep intronic and other noncoding WGS variants will improve, thereby increasing the diagnostic advantage of WGS over WES. Our prediction, therefore, is that the perceived disadvantages of WGS relative to WES, including increased cost and increased requirements for data analysis and storage, will ultimately be outweighed by its diagnostic superiority.
Study advantages and limitations
Our study employed prospective recruitment from multiple pediatric subspecialty clinics where NGS multigene panel testing is standard of care. Although phenotypes were highly variable, referrals were disproportionately from three clinics. For example, there were only three referrals from the Neurology clinic, despite a majority of participants having a neurologic phenotype. There are several potential contributing factors: (i) considerable overlap among patients seen in the Neurology, Ophthalmology, and (Neuro-)Metabolic clinics, (ii) differences in comfort level of individual health-care providers at The Hospital for Sick Children with respect to clinical genetic testing (e.g., highest on average among our metabolic geneticist colleagues), and (iii) enrollment of patients in alternative, clinic-specific genetic research studies.
We were underpowered to test for additional factors potentially influencing diagnostic yield in this heterogeneous study population. For example, we were unable to robustly investigate the degree to which involvement of a clinical geneticist contributed to a diagnosis via conventional testing. It is also possible that a patient’s participation in this study affected the clinician’s genetic testing strategy. An alternative (trio-based) study design that facilitates prioritization of de novo variants may result in increased diagnosis via WGS. Analysis and discussion of secondary findings is beyond the scope of this report, but represents an important consideration in the clinical use of WES and WGS.31 Gene discovery was also not the focus of this study.
Neither the WGS nor the WES performed in this study were clinically validated tests, but all diagnostic variants were clinically confirmed prior to their return to participants. There are some inherent challenges in comparing a research-based test (WGS) with clinical genetic testing. Cost, timing of obtaining results, and scalability are specific considerations.8 This study did not quantify the time associated with the interpretation of WGS results, which represent orders of magnitude more data than most traditional tests. However, time to results is expected to steadily improve with advancements in algorithmic pipelines and public database variant curation. The often-quoted (low) cost of WGS can be misleading because it does not take into account extended bioinformatics, variant interpretation, overhead costs, and pre- and post-test genetic counseling associated with providing WGS-based testing as a clinical service. This study was not designed to directly compare costs of WGS with standard-of-care testing. Some individuals had already had some genetic testing prior to enrollment in this study. Nonetheless, the high cumulative cost of iterative testing in this study is consistent with data from comparative cost studies involving WES and conventional testing,29, 32 and there are reasons to expect the value of WGS to increase over time.33
Future directions
These prospective data provide new insights into how WGS could transform genetic assessment in pediatric medicine. As clinical WGS becomes feasible on a larger scale, it may ultimately become a first-tier diagnostic test. Hypothesis-driven testing would still be performed, by limiting the initial analysis of WGS data to specific genes and loci. Provided coverage is adequate, this would allow the clinician to retain control over the scope of testing, while also facilitating future comprehensive interrogation of the genome. Further research will help to delineate the potential advantages and limitations of such an approach.
References
Stavropoulos DJ, Merico D, Jobling R et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom Med 2016;1:15012.
Winkler EC, Wiemann S . Findings made in gene panel to whole genome sequencing: data, knowledge, ethics—and consequences? Expert Rev Mol Diagn 2016;16:1259–1270.
Lee H, Deignan JL, Dorrani N et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880–1887.
Posey JE, Harel T, Liu P et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017;376:21–31.
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017;25:176–182.
Stark Z, Tan TY, Chong B et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016;18:1090–1096.
Gilissen C, Hehir-Kwa JY, Thung DT et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511:344–347.
Soden SE, Saunders CJ, Willig LK et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014;6:265ra168.
Taylor JC, Martin HC, Lise S et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 2015;47:717–726.
Willig LK, Petrikin JE, Smith LD et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med 2015;3:377–387.
Bowdin SC, Hayeems RZ, Monfared N et al. The SickKids Genome Clinic: developing and evaluating a pediatric model for individualized genomic medicine. Clin Genet 2016;89:10–19.
Kalia SS, Adelman K, Bale SJ et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249–255.
Girdea M, Dumitriu S, Fiume M et al. PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat 2013;34:1057–1065.
Raczy C, Petrovski R, Saunders CT et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 2013;29:2041–2043.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
Castellana S, Ronai J, Mazza T . MitImpact: an exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum Mutat 2015;36:E2413–2422.
Zhu M, Need AC, Han Y et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet 2012;91:408–421.
Abyzov A, Urban AE, Snyder M, Gerstein M . CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011;21:974–984.
Landrum MJ, Lee JM, Benson M et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–868.
Stenson PD, Ball EV, Mort M et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 2003;21:577–581.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Consugar MB, Navarro-Gomez D, Place EM et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med 2015;17:253–261.
Makrythanasis P, Kato M, Zaki MS et al. Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. Am J Hum Genet 2016;98:615–626.
Merico D, Roifman M, Braunschweig U et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat Commun 2015;6:8718.
Pengelly RJ, Greville-Heygate S, Schmidt S et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet 2016;53:735–742.
Engel AG, Selcen D, Shen XM et al. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet 2016;2:e105.
Tammimies K, Marshall CR, Walker S et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 2015;314:895–903.
Wenger AM, Guturu H, Bernstein JA, Bejerano G . Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med 2017;19:209–214.
Stark Z, Schofield D, Alam K et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med; e-pub ahead of print 26 January 2017.
Lelieveld SH, Spielmann M, Mundlos S et al. Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum Mutat 2015;36:815–822.
Anderson JA, Meyn MS, Shuman C et al. Parents perspectives on whole genome sequencing for their children: qualified enthusiasm? J Med Ethics; e-pub ahead of print 25 November 2016.
Monroe GR, Frederix GW, Savelberg SM et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med 2016;18:949–956.
Tsiplova K, Zur RM, Marshall CR et al. A microcosting and cost-consequence analysis of clinical genomic testing strategies in autism spectrum disorder. Genet Med; e-pub ahead of print 4 May 2017.
Vetrini F, Tammaro R, Bondanza S et al. Aberrant splicing in the ocular albinism type 1 gene (OA1/GPR143) is corrected in vitro by morpholino antisense oligonucleotides. Hum Mutat 2006;27:420–426.
Ogino W, Takeshima Y, Nishiyama A et al. Mutation analysis of the ornithine transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe J Med Sci 2007;53:229–240.
Engel K, Nuoffer JM, Muhlhausen C et al. Analysis of mRNA transcripts improves the success rate of molecular genetic testing in OTC deficiency. Mol Genet Metab 2008;94:292–297.
Hinkes B, Wiggins RC, Gbadegesin R et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006;38:1397–1405.
Ng BG, Buckingham KJ, Raymond K et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet 2013;92:632–636.
Heinen CA, Jongejan A, Watson PJ et al. A specific mutation in TBL1XR1 causes Pierpont syndrome. J Med Genet 2016;53:330–337.
Lionel AC, Monfared N, Scherer SW et al. MED23-associated refractory epilepsy successfully treated with the ketogenic diet. Am J Med Genet A 2016;170:2421–2425.
Acknowledgements
This study was funded by the Centre for Genetic Medicine, The Centre for Applied Genomics, The Hospital for Sick Children, Genome Canada, and the University of Toronto McLaughlin Centre. A.C.L. was supported by a CREMS Research Scholarship from the Faculty of Medicine and McLaughlin Centre at the University of Toronto. S.W.S. holds the Canadian Institutes for Health Research (CIHR) GlaxoSmithKline Endowed Chair in Genome Sciences at The Hospital for Sick Children and the University of Toronto. R.D.C. holds the Women’s Auxiliary Chair in Clinical and Metabolic Genetics at The Hospital for Sick Children. The authors thank the patients and families whose participation made this project possible, the many health-care providers involved in their care, and staff at The Centre for Applied Genomics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The first three authors contributed equally to this work.
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lionel, A., Costain, G., Monfared, N. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 20, 435–443 (2018). https://doi.org/10.1038/gim.2017.119
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.119
Keywords
This article is cited by
-
Recent advances in polygenic scores: translation, equitability, methods and FAIR tools
Genome Medicine (2024)
-
Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders
Human Genomics (2024)
-
The expanding diagnostic toolbox for rare genetic diseases
Nature Reviews Genetics (2024)
-
RNA sequencing and target long-read sequencing reveal an intronic transposon insertion causing aberrant splicing
Journal of Human Genetics (2024)
-
Combining a prioritization strategy and functional studies nominates 5’UTR variants underlying inherited retinal disease
Genome Medicine (2024)
