
Abstract
Purpose
Based on prenatal suspicion of the combination of radioulnar or radiohumeral synostosis and a peculiar shape of the skull suggestive of craniosynostosis, we report on six patients from four unrelated consanguineous families in whom Antley-Bixler syndrome was suspected during the prenatal period without mutation in genes known to be associated with the syndrome.
Methods
Molecular diagnosis involved whole-exome and gene-panel sequencing.
Results:
All sequenced patients showed a unique homozygous mutation of c.667G>A, p.Gly223Ser (NM_012200) in the beta-1,3-glucuronyltransferase 3 (B3GAT3) gene known to be involved in linkeropathy syndrome. Linkeropathies correspond to a recently identified group of heterogeneous genetic syndromes along a spectrum of skeletal and connective tissue disorders. These patients featured mainly craniosynostosis, midface hypoplasia, bilateral radioulnar synostosis, multiple neonatal fractures, dislocated joints, joint contracture, long fingers, foot deformity, and cardiovascular abnormalities. All died before 1 year of age.
Conclusion
We identified a novel B3GAT3-related disorder with craniosynostosis and bone fragility, due to a unique homozygous mutation in B3GAT3. This syndrome should be considered in the prenatal period in light of the severe outcome and as an alternative diagnosis to Antley-Bixler or Shprintzen-Goldberg syndrome.
Similar content being viewed by others


Introduction
Linkeropathies correspond to a recently identified group of heterogeneous genetic syndromes due to mutations in genes involved in proteoglycan (PG) synthesis pathway. PGs are ubiquitous components of extracellular matrix, subdivided into chondroitin sulfate, dermatan sulfate, and heparan sulfate PGs. They are fundamental for the development of several organs including skeletal muscle, bones, cartilage, retina, and heart.1 Linkeropathy genes encode for enzymes in charge of branching glycosaminoglycan onto PG via a tetrasaccharide linker. Five genes have been identified so far: two xylotransferases, namely xylosyltransferase 1 (XYLT1, OMIM 608124) and xylosyltransferase 2 (XYLT2, OMIM 608125); two galactosyltransferases, namely xylosylprotein 4-beta-galactosyltransferase polypeptide 7 (B4GALT7, OMIM 604327) and beta-1,3-galactosyltransferase polypeptide 6 (B3GALT6, OMIM 615291); and one glucuronyltransferase, beta-1,3-glucuronyltransferase 3 (B3GAT3, OMIM 606374). Mutations affecting these genes are responsible for a spectrum of skeletal and connective tissue disorders combining multiple congenital contracture and/or dislocation, joint laxity, broad digits, brachycephaly, small mouth, prominent eyes due to glaucoma, short or webbed neck, heart malformation, and developmental delay.2, 3 XYLT1 and XYLT2 mutations are associated respectively with Desbuquois dysplasia type 2 (OMIM 615777) and with spondylo-ocular syndrome (OMIM 605822). B4GALT7 mutations are described in a form of Elhers-Danlos syndrome characterized by short stature and limb anomalies (OMIM 130070). B3GALT6 mutations are involved in two genetic disorders: spondyloepimetaphyseal dysplasia with joint laxity type 1 (OMIM 271640) and Ehlers-Danlos syndrome progeroid type 2 (OMIM 615349). Finally, B3GAT3 mutations are reported with multiple joint dislocations, short stature, and craniofacial dysmorphism with or without congenital heart defects (OMIM 245600).
We performed next-generation sequencing for five of six patients from four unrelated families in whom Antley-Bixler syndrome (ABS1, OMIM 201750 and ABS2, OMIM 207410) was suspected during the prenatal period based on the combination of radioulnar or radiohumeral synostosis and a peculiar shape of the skull suggestive of craniosynostosis, where no mutations in known genes involved in ABS were reported.
Materials and methods
Clinical data
We identified six patients and obtained written informed consent for genetic testing and DNA sampling for five of them and their relatives. Specific written informed consent for the publication of photos was obtained for three patients. All parents were healthy during pregnancy. They were all natives of Morocco and were first cousins. Follow-up of the six infants occurred at Montpellier and Nîmes hospitals. ABS was suspected from ultrasonography and CT-scan findings. Molecular studies (except for patient 4) of cytochrome P450 oxidoreductase gene (POR, OMIM 124015) and fibroblast growth factor receptor 2 gene (FGFR2, OMIM 176943) showed no mutations. Clinical data are summarized in Table 1 and full clinical descriptions are in Supplementary Data S1 online.
Whole-exome sequencing
DNA from patient 1 from family 1 and her mother was analyzed by whole-exome sequencing at the Centre National de Génotypage (Institut de Génomique, CEA, Évry, France). After complete DNA quality control for each sample (duplicate quantification, evaluating DNA integrity/quality, checking for absence of PCR inhibitors), genomic DNA (3 μg) was captured using an in-solution enrichment method (Human All Exon v5—50 Mb, Agilent Technologies, Santa Clara, CA). Library preparation and exome enrichment (~20.000 targeted genes) were performed automatically using NGSx (PerkinElmer, Waltham, MA) and Bravo (Agilent Technologies), respectively, according to the manufacturer’s instructions (SureSelect, Agilent Technologies). After normalization and quality control, exome-enriched libraries were sequenced using the Illumina HiSeq2000 system (Illumina, San Diego, CA) as paired-end 100-bp reads. Samples were sequenced as pools of four samples per lane, to obtain an average coverage of 70 to 80 X, with at least 80% of the target nucleotides covered at 30 X. Image analysis and base calling involved use of the Illumina Real Time Analysis Pipeline. Sequence-quality parameters were assessed daily during the 12 days of sequencing. The standard bioinformatics analysis of sequencing data was based on the Illumina pipeline (CASAVA1.8.2) to generate a FASTQ file for each sample.
Gene-panel sequencing
DNA samples from patients 2, 3, and 5 underwent gene-panel sequencing for a constitutional bone-disease gene panel of 49 genes, including linkeropathy genes (B3GAT3, B3GALT6, B4GALT7, XYLT1, and XYLT2) and ABS genes (POR, FGFR2 and CYP26B1). Library enrichment was performed with SureSelectQXT Target Enrichment for Illumina Multiplexed Sequencing, Version C0, January 2015 (Agilent Technologies), according to the manufacturer’s protocol.
Enriched library sample sequencing involved the Illumina MiSeq sequencer (Illumina, San Diego, CA) with the MS-103-1002 MiSeq Reagent Micro Kit, v2 (300 cycles). FASTQ files from patient samples were loaded into the MiSeq Reporter (v2.2.31), and reads were mapped against the genomic sequences (human genome version hg19) of the targeted genes. For secondary data analysis, BAM files of samples were loaded into the SeqPilot SeqNext module (v4.0, JSI Medical Systems, Kippenheim, Germany).
Sanger sequencing
Exon 3 of B3GAT3 gene underwent direct Sanger sequencing to confirm next-generation sequencing results as described (BDT3.1 technology in a ABI3130XL sequencer; Barat-Houari et al., 20154) on a DNA sample from probands and their relatives, except for patient 4, because neither DNA samples nor informed consent were available.
Results
Clinical data
Ultrasonography at the second trimester of pregnancy revealed several malformations in our six patients, namely restricted hand movement (n = 4), equinovarus position of feet (n = 4), hypoplasia of nasal bone (n = 3), ventricular septal defect (n = 2), overlapping fingers (n = 2) and suspicion of bilateral radioulnar synostosis (n = 2) (Figure 1).

Prenatal investigation. (a) 3D ultrasonography reconstruction in patients 3 (front view) and 4 (lateral view, hand and feet). Note midface hypoplasia, clenched hand and varus feet. (b) Prenatal ultrasonography of patient 1, computed tomography scan, and 3D reconstruction showing bilateral proximal radioulnar synostosis (arrows).
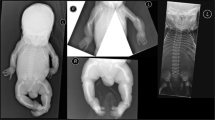
In the postnatal period, patients mainly presented bilateral radioulnar synostosis (6/6), joint contracture (6/6), foot deformity (6/6), midface hypoplasia (5/6), multiple neonatal fractures (4/6), abnormally long fingers (4/6), dislocated joints (3/5), craniosynostosis (3/5), and cardiovascular abnormalities (3/6) (Figure 2). All patients died before the age of one year. Clinical features are fully described in Table 1 and Supplementary data S1.

Clinical and radiological features in reported patients. Postnatal clinical data: front, lateral and extremities view in patients 1 (a), 5 (b), and 6 (c). Note clenched hands, varus feet, arthrogryposis, midface hypoplasia, and brachycephaly, as well as ocular proptosis in some patients. Radiological features: skeletal X-ray of patient 2 showing (d) fracture of right femur, (e) radioulnar synostosis, and (f) finger deformations. 3D reconstruction and computed tomography scan showing coronal and lambdoid craniosynostosis of patients 3 (g) and 4 (h).
Exome sequencing
Post-natal exome analysis of patient 1 revealed a potentially pathogenic homozygous variation, c.667G>A, p.Gly223Ser, in exon 4 of B3GAT3 (NM_012200). The homozygous c.667G>A mutation was confirmed by Sanger sequencing in patient 1 and segregation analysis revealed that both parents were heterozygous for this variation. According to the ExAC database , this mutation was already reported in 1/119,290 alleles, with a frequency of 0.000008. This variant in ExAC was reported in a European (non-Finnish) population. Nucleotide and amino-acid multiple alignments among different animal species revealed that p.Gly223 is highly conserved. In silico variant prediction tools with wANNOVAR5 included SIFT T (0.07), PolyPhen2 HDIV D (0.999), and Combined Annotation Dependent Depletion (phred score 33). No genetic variation was observed in known genes involved in ABS or an ABS-like phenotype, nor in the other linkeropathy genes. This variant was reported once in the literature for one patient with a linkeropathy diagnosis.3
Gene-panel sequencing
Analysis with a gene panel approach for three other probands, patients 2, 3, and 5, revealed the same homozygous mutation, c.667G>A, p.Gly223Ser, in B3GAT3 (NM_012200) identified by whole-exome sequencing in patient 1. Sanger sequencing confirmed that affected patients were indeed homozygous for the mutation. Segregation analysis revealed that all parents were heterozygous for the same mutation.
Discussion
Using exome and gene-panel sequencing, we characterized a unique homozygous mutation, c.667G>A, p.Gly223Ser, in B3GAT3 in five of our six patients. Recently, a Mexican boy with this same mutation was described.3 The association of a similar phenotype for this patient, namely, radioulnar or radiohumeral synostosis, midface hypoplasia, and joint contracture, led us to consider B3GAT3 as the gene responsible for the phenotype in our patients. In contrast to other linkeropathy genes encoding for xylosyltransferase or galactosyltransferase, B3GAT3 is the only one to encode for a glucuronosyltransferase, an enzyme involved in post-translational modification and assembly of PG. Conversely, this enzyme links glycosaminoglycans to the core protein of PGs by the addition of glucuronic acid, the last component of the linker region between glycosaminoglycans and PGs. Gulberti et al.6 demonstrated that substituting an alanine residue for Gly222 or Gly223 reduced the glucuronosyltransferase 1 activity of recombinant human glucuronosyltransferase 1 in yeast cells. The Gly223 mutation is localized in the catalytic acceptor substrate-binding subdomain (residues 198–308, Pedersen et al.7). Baasenjav et al.8 described patients with a mutation in this subdomain and B3GAT3 expression shown in mice hearts, bones, aortas, and osteoblasts. The clinical features they share with our patients suggest that p.Gly223Ser mutations are pathogenic. Table 1 summarizes the clinical manifestations of patients with homozygous B3GAT3 c.667G>A mutation from the literature and our patients. We contemplated a unique common ancestor for all patients, but the fact that the patients with the same rare homozygous mutation were from different countries and continents led us to consider a “hotspot” mutation in B3GAT3. We suggest that this B3GAT3-related disorder with craniosynostosis and bone fragility presentation should be considered as a differential diagnosis in the prenatal period for ABS and in the postnatal period for Shprintzen-Goldberg syndrome (SGS, OMIM 182212). Indeed ABS is suspected during pregnancy, especially during the second and third trimester, if ultrasonography provokes a strong suspicion of craniofacial deformities, based on the suspicion of craniosynostosis (a hallmark of ABS), midface hypoplasia, bilateral radiohumeral or radioulnar synostosis, and femoral bowing.9, 10, 11 We reviewed the literature for ABS patients and found a patient12 without a molecular diagnosis but with a strikingly similar phenotype: craniosynostosis, midface hypoplasia with proptosis, long tapered fingers, elbow joint contracture due to radioulnar synostosis, and equinovarus position of feet. Without mutation of the FGFR2 gene on molecular analysis, we suggest searching for the B3GAT3 c.667G>A mutation in this patient. In the postnatal period, SGS could also be suspected, based on the association of craniosynostosis and arachnodactyly. Indeed, SGS is characterized by craniosynostosis and distinctive craniofacial features, skeletal anomalies (arachnodactyly, dolichostenomelia, camptodactyly, pes planus, pectus excavatum or carinatum, scoliosis, joint hypermobility or contractures, and C1/C2 spine malformation), cardiovascular anomalies, intellectual deficiency, neurologic and brain anomalies (hydrocephalus, dilatation of the lateral ventricles, and Chiari 1 malformation). The SKI proto-oncogene (SKI, OMIM 182212) is the only gene related to SGS known to date.13
B3GAT3 is involved in a spectrum of several connective tissue and skeletal disorders. Table 2 summarizes the clinical features in all patients with the B3GAT3 mutation. Among the different B3GAT3-related disorders, a Larsen-like phenotype characterized by joint dislocation and congenital heart defect was reported in three different studies. Von Oettingen et al.14 reported an undetermined syndrome in a 5-year-old boy presenting skeletal dysplasia with bilateral radioulnar synostosis, brachycephalia, global developmental delay, and multiple congenital anomalies. Baasanjav et al.8 described patients with short stature, radioulnar synostosis, exophthalmia, cardiac abnormalities, and brachycephaly. All patients had the same homozygous missense mutation in B3GAT3 (c.830G>A, p.Arg277Gln). In a second large consanguineous Indonesian family, Budde et al.15 described eight patients with bone dysplasia and short stature. The patients also had dislocated joints with short upper arms, midface hypoplasia, and broad fingers and toes. No cardiac involvement was described. All patients had the same homozygous missense mutation in B3GAT3 (c.419C>T, p.Pro140Leu).
Alazami et al.16 reported a gerodermia osteodysplatica–like syndrome in a patient with spondyloepimetaphyseal dysplasia, cutis laxa, osteoporosis, fracture, multiple bony chondromas, and short stature. The patient had a homozygous missense mutation in B3GAT3 (c.245C>T, p.Pro82Leu). Phenotypes of the linkeropathy gene B3GAT3 are heterogeneous but precise B3GAT3-related disorder (B3GAT3-RD) phenotype seems to be related to specific mutations (Figure 3).

Besides the observation of a presumed well-separated phenotype, a phenotypic continuum could be considered in light of the description of a 6-year-old boy17 who presented hip-joint dislocation due to severe ligamentous laxity, osteoporosis, kyphoscoliosis, multiple fractures, and cardiac abnormalities (patent foramen ovale, bicuspid aortic valve, mild aortic root enlargement, and ascending aorta dilation). The patient had a compound mutation in B3GAT3 (c.1A>G, p.Met1?; c.671T>A, p.Leu224Gln). Existence of an intermediate phenotype with compound mutation led us to discuss also the possibility of a continuum phenotype classification for B3GAT3-RD.
Here we report a novel B3GAT3-RD with craniosynostosis and bone fragility. We describe an expanded craniofacial phenotype of linkeropathy syndromes, especially craniosynostosis, and six patients with c.667G>A B3GAT3 homozygous mutation. This gene should be screened, along with POR, FGFR2, and CYP26B1 (OMIM 605207) in patients with suspected ABS during the prenatal period, because of the lethal severity of symptoms in the early months of life. This B3GAT3-RD should be suspected in the postnatal period as an alternative diagnosis to ABS or SGS.
References
Taylan F, Mäkitie O . Abnormal proteoglycan synthesis due to gene defects causes skeletal diseases with overlapping phenotypes. Horm Metab Res 2016;48:745–754.
Nakajima M, Mizumoto S, Miyake N et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet 2013;92:927–934.
Jones KL, Schwarze U, Adam MP, Byers PH, Mefford HC . A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am J Med Genet A 2015;167:2691–2696.
Barat-Houari M, Dumont B, Fabre A et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet 2016;24:992–1000.
Chang X, Wang K . wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 2012;49:443–436.
Gulberti S, Lattard V, Fondeur M et al. Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human B1,4-galactosyltransferase 7 (GalT-I) and B1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage. J Biol Chem 2005;280:1417–1425.
Pedersen LC, Darden TA, Masahiko Negishi M . Crystal structure of B1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J Biol Chem 2002;277:21869–21873.
Baasanjav S, Al-Gazali L, Hashiguchi T et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet 2011;89:15–27.
Delahaye S, Bernard JP, Rénier D, Ville Y . Prenatal ultrasound diagnosis of fetal craniosynostosis. Ultrasound Obstet Gynecol 2003;21:347–353.
Oldani E, Garel C, Bucourt M, Carbillon L . Prenatal diagnosis of Antley-Bixler syndrome and POR deficiency. Am J Case Rep 2015;16:882–885.
Machado LE, Osborne NG, Bonilla-Musoles F . Antley-Bixler syndrome: report of a case. J Ultrasound Med 2001;20:73–77.
Crisponi G, Porcu C, Piu ME . Antley-Bixler syndrome: case report and review of the literature. Clin Dysmorphol 1997;6:61–68.
Greally MT. Shprintzen-Goldberg Syndrome. 2006 Jan 13 [Updated 2013 Jun 13]. In: Pagon RA, Adam MP, Ardinger HH et al (eds). GeneReviews. (WA): University of Washington: : Seattle, WA, 1993-2017. https://www.ncbi.nlm.nih.gov/books/NBK1277/. Accessed March 2017.
Oettingen JE, von, Tan W-H, Dauber A . Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy–report of the second family with B3GAT3 mutation and expansion of the phenotype. Am J Med Genet A 2014;6:1580–1586.
Budde BS, Mizumoto S, Kogawa R et al. Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel mutation in B3GAT3. Hum Genet 2015;134:691–704.
Alazami AM, Al-Qattan SM, Faqeih E et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum Genet 2016;135:525–540.
Job F, Mizumoto S, Smith L et al. Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: expanding the disease phenotype. BMC Med Genet 2016;17:86.
Murzin AG, Brenner SE, Hubbard T, Chothia C . SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 1995;247:536–540.
Berman HM, Westbrook J, Feng Z et al. The Protein Data Bank. Nucl Acid Res 2000;28:235–242.
Acknowledgements
Part of this work was supported by the Programme Hospitalier de Recherche Clinique Regional (no. 7855) and the French Health Ministry (Direction Générale de l’Organisation des Soins–DGOS). This work was performed with the support of the High Performance Computing Platform HPC@LR, financed by the Occitanie/Pyrénées-Méditerranée Region, Montpellier Mediterranean Metropole, and the University of Montpellier. We thank the families for their participation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The first two authors contributed equally to this work.
Supplementary material is linked to the online version of the paper at
Rights and permissions
About this article

Cite this article
Yauy, K., Tran Mau-Them, F., Willems, M. et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet Med 20, 269–274 (2018). https://doi.org/10.1038/gim.2017.109
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.109
Keywords
This article is cited by
-
Severe phenotypes of B3GAT3-related disorder caused by two heterozygous variants: a case report and literature review
BMC Medical Genomics (2022)
-
The clinical and mutational spectrum of B3GAT3 linkeropathy: two case reports and literature review
Orphanet Journal of Rare Diseases (2019)
