
Abstract
Patients with amyotrophic lateral sclerosis (ALS) often have questions about why they developed the disease and the likelihood that family members will also be affected. In recent years, providing answers to these questions has become more complex with the identification of multiple novel genes, the newly recognized etiologic link between ALS and frontotemporal dementia (FTD), and the increased availability of commercial genetic testing. A genetic diagnosis is particularly important to establish in the era of emerging gene-based therapies, such as SOD1 antisense oligonucleotide trials. In the span of a few years, ALS genetic testing options have progressed from testing of a single gene to multigene next-generation sequencing panels and whole-exome sequencing. This article provides suggestions for genetic counseling and genetic testing for ALS in this new environment.
Genet Med 19 3, 267–274.
Similar content being viewed by others
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disorder characterized by loss of upper and lower motor neurons, progressive paralysis, and death within an average of 2–5 years after symptom onset. Diagnosis is based on clinical features, electrodiagnostic testing, and exclusion of other diseases with overlapping symptoms. Treatment is palliative, and there are currently no effective disease-modifying medical therapies other than riluzole, which slows progression and prolongs survival by an average of 3 months.1 Although the majority of cases appear to occur as sporadic ALS (sALS), approximately 5–10% of ALS patients have a family history of ALS (familial ALS (fALS)).2,3 Clinically, fALS and sALS are similar. However, fALS cases are distinguished by an earlier mean age of onset (46 years) than sALS cases (56 years).4,5 Disease progression in fALS may be significantly shorter or longer than in sALS and may be associated with specific genetic mutations.4,6 The distinction between fALS and sALS is increasingly blurred as common biologic and genetic features are recognized, although classifying ALS cases as familial or sporadic remains useful in clinical practice.7
Patients with ALS often ask why they developed the disease, what the chance is that they will pass it on to their children, and how quickly their condition will progress.8,9 Historically, the majority of patients (with sALS) were reassured that no one else in their family was at risk. Patients with fALS were told that their condition could be passed on to children, but because genetic testing was limited to SOD1, accounting for 20% of fALS cases, most patients had to accept that the precise cause of their condition could not be determined.10
In recent years, the landscape of genetic testing and genetic counseling for ALS has been transformed with the identification of novel genes, including C9orf72, the recognition of the link between ALS and frontotemporal dementia (FTD), and the advent of next-generation sequencing technology. The genetic basis of two-thirds of fALS and 10% of sALS case has now been established. The 2011 discovery of the hexanucleotide repeat expansion in C9orf72 led to the identification of pathogenic expansions in a significant percentage of both fALS and sALS and revealed a common genetic etiology of ALS and FTD.11,12 Although ALS patients desire access to genetic testing,13 genetic advances have been slow to reach the clinical care of the ALS patient.10
In this article, we review the current understanding of ALS genetics, outline genetic counseling considerations, and summarize testing options and factors to consider when determining appropriate genetic testing for ALS patients. This information may be useful to geneticists, genetic counselors, neurologists, and other clinicians who provide care and education to patients with ALS. With increased access to genetic testing and counseling, ALS patients and families may benefit from the many recent advances in the field of ALS genetics.
Overview of ALS Genetics
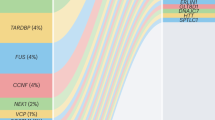
Study of fALS pedigrees has led to the discovery of more than 25 ALS genes,14 which account for two-thirds of fALS cases and approximately 11% of sALS cases7 (see Figure 1 ). The majority of ALS genes are inherited in a dominant fashion with variable and age-dependent penetrance. Significant intrafamilial and interfamilial variability in age of onset and disease progression is observed.15 Genes that may cause recessive disease include OPTN, SPG11, FUS, and SOD1 (specifically, the homozygous Asp90Ala mutation), among others; the UBQLN2 gene is X-linked dominant. Here, we review the most prevalent ALS genes and highlight several minor genes associated with unique “crossover” motor neuron disease phenotypes.

The proportion of amyotrophic lateral sclerosis cases attributable to each gene in populations of European descent.
C9orf72. A pathogenic hexanucleotide (G4C2) repeat expansion in the first intron of C9orf72 accounts for approximately 40% of fALS and approximately 7% of sALS, approximately 25% of familial FTD,7,16 and up to 88% of cases of combined ALS/FTD.17 These frequencies far exceed those of any other ALS or FTD gene; however, pathogenic C9orf72 repeat expansion frequencies vary greatly by ethnicity/geographic origin. The highest frequencies are reported in northern European (particularly Scandinavian) countries, with low frequencies reported in Asian countries.17 To date, the C9orf72 repeat expansion is the only ALS mutation found at >2% frequency in sALS.7
In addition to the upper and lower motor neuron dysfunction characteristic of ALS, the C9orf72 repeat expansion causes frontotemporal lobar degeneration, manifesting clinically with behavior changes, executive dysfunction, and/or language impairment. Brain MRI may show symmetric bilateral frontal lobe atrophy.17 On FDG PET, ALS patients with C9orf72 expansions may show hypometabolism in the anterior and posterior cingulate cortex, insula, caudate, and thalamus, and the left frontal and superior temporal cortex, and hypermetabolism in the midbrain, bilateral occipital cortex, globus pallidus, and left interior temporal cortex compared to patients with sporadic ALS.18 In addition to ALS and FTD, C9orf72 expansions may cause parkinsonism (typically the akinetic rigid type without tremor19) and psychotic symptoms.20 Age at onset is most often between 30 and 70 years. The first symptoms may be solely FTD or ALS, with additional symptoms developing with disease progression.21
The size of the G4C2 repeat in C9orf72 alleles ranges from 2 repeats to more than 4,000 (refs. 11,12); disease alleles are characterized by repeat expansion. Repeat alleles of <25 G4C2 units are typically considered normal.16,22 The repeat size at which pathogenicity occurs is not precisely clear. As outlined by Gijselnick et al.,23 establishing a clear repeat size cutoff for pathogenicity has been complicated by the identification of intermediate size alleles of 25–60 repeat units in FTD and ALS (but not consistently segregating with disease) and variable laboratory methods and imprecision in sizing larger repeats. Additionally, intraindividual variation of repeat sizes may be significant in expansion carriers, with larger repeat sizes occurring in neural compared to nonneural tissues.24 Although there are literature reports of anticipation, this has not been clearly established as a feature of C9orf72 ALS/FTD.7,23
The C9orf72 repeat expansion is transmitted in an autosomal-dominant fashion, with variable and age-dependent penetrance. Most affected individuals have an affected parent. However, lack of knowledge of family history, lack of recognition of disease in a parent, death of a parent before symptom onset, and other circumstances can lead to an apparently negative family history. Early studies have estimated cumulative age-dependent penetrance of 0% at age 35 years, 50% at 58 years, and almost 100% by 80 years.16,25 However, neurologically healthy and elderly obligate carriers of disease alleles are known to occur in ALS/FTD families, and expanded repeat alleles have been identified in 0.2–0.6% of unaffected population controls, including the elderly.21 Further study is needed to understand the penetrance of the C9orf72 repeat expansion. Preclinical efforts are currently underway to investigate the use of allele-specific oligonucleotides (ASOs) to treat the pathologic features of the C9orf72 expansion.26
SOD1. SOD1 was the first gene reported as a cause of fALS and was identified via linkage analysis.27 Mutations in SOD1 are the second most common known cause of fALS after C9orf72, and they are reported to occur in approximately 12% of fALS and approximately 1–2% of sALS cases.28 The frequency of SOD1 mutations varies widely by population, occurring in 38% of fALS cases in Iran29 and 0% in Ireland.30 Clinically, SOD1 mutations are usually not associated with cognitive impairment, and bulbar onset is less common than in other fALS types.31 Some genotype–phenotype correlations are known; the Ala5Val mutation (denoted in older reports as Ala4Val) accounts for half of SOD1 mutations in North America and causes a rapidly progressive course and death within 1 year of symptom onset.6 Other mutations are associated with a prolonged disease course of 10 years or more, including Gly37Arg, Gly41Asp, His46Arg, Glu100Lys,4,6 and Asp90Ala.32
SOD1 mutations are typically inherited in an autosomal-dominant manner. The Asp90Ala mutation, however, is recessive in northern Scandinavia33 and may be dominant in other populations.34 Dominant SOD1 mutations are associated with variable and age-dependent penetrance. Penetrance figures may overestimate risk because of ascertainment bias of families with high penetrance mutations. In a large pedigree with the Ile113Thr mutation, penetrance was estimated as 50% by age 60 years and 88% by age 80 years.35
A phase I trial of low doses of a therapeutic oligonucleotide targeting SOD1 was completed in 2012 with no adverse effects compared to placebo.26 Several clinical trials are currently investigating therapies in SOD1 mutation carriers (clinicaltrials.gov NCT01083667, NCT00706147, and NCT02623699).
TARDBP. Mutations in TARDBP are dominantly transmitted and found in up to approximately 4% of fALS and approximately 1% of sALS cases.36 Prior to the discovery of mutations in this gene in fALS, its protein product (TDP-43) was found to be a component of the ubiquitin-positive neuronal inclusions characteristic of both ALS and FTD.37 TARDBP mutations have been identified in geographically diverse ALS families; a founder effect in Sardinia is associated with a high frequency of the Ala382Thr mutation.36 TARDBP-associated ALS is considered to have a typical clinical presentation that may involve FTD and, rarely, FTD without motor neuron disease.38,39
FUS. FUS mutations are reported to account for approximately 4% of fALS and approximately 1% of sALS cases overall,7,40,41 with the highest frequency (8.7%) identified in German fALS cases.42 Recurrent mutations identified in multiple families include Arg521Cys and Arg521Gly.43 Review of clinical information for 20 affected individuals revealed even gender distribution, average age of onset of 44.5 years, and cervical onset in 10, lumbar onset in five, and bulbar onset in three.41 FUS mutations have been reported in FTD.44 FUS mutations have been associated with a shorter life span, although extensive intrafamilial variability has been observed.31 Truncating mutations have been identified in patients with early onset and an aggressive disease course.42 Recently, de novo FUS mutations were shown to be the most frequent cause of early-onset ALS (onset at age younger than 35 years) in a German series.45 FUS mutations are typically dominant, although recessive inheritance of the homozygous His517Gln mutation has been reported in a fALS family of Cape Verdean origin.40
VCP. Mutations in VCP were first reported in families with inclusion body myopathy, Paget disease, and frontotemporal dementia, and were later recognized as a cause of fALS.46 Mutations in VCP may lead to a range of clinical presentations, even within the same family: ALS, ALS-FTD, ALS with Paget’s disease, and inclusion body myopathy and FTD.46,47,48 VCP mutations are dominantly transmitted; the percentage of fALS cases attributable to VCP is estimated at 1–2%.46,48
MATR3. A missense mutation in MATR3, Ser85Cys, was first reported in two families with autosomal-dominant distal myopathy49; different missense mutations were later found in patients with sALS and fALS.50 Re-examination of one of the originally reported families identified brisk reflexes in some individuals, indicating motor neuron pathology. Individuals in that family developed progressive respiratory failure resulting in death, typically 15 years after symptoms onset, whereas other MATR3 mutations have been associated with a more typical ALS progression.50 MATR3 mutations are dominantly transmitted and appear to be a rare cause of ALS.50,51
CHCHD10. The Ser59Leu mutation in CHCHD10, a nuclear gene encoding a small mitochondrial protein, was first reported in a large family with late-onset motor neuron disease, cognitive decline resembling FTD, cerebellar ataxia, and myopathy. Muscle biopsy specimens of affected individuals showed ragged-red and cytochrome c oxidase–negative fibers with combined respiratory chain deficiency and abnormal assembly of complex V.52 Other mutations have been subsequently reported in ALS, ALS-FTD, and FTD patients.53,54,55,56,57,58 CHCHD10 mutations are dominantly transmitted and appear to account for <1% of ALS cases.59
Genetic Counseling and Genetic Testing
Genetic risk assessment
As with all clinical genetic evaluations, family history is a fundamental component of ALS genetic risk assessment and genetic counseling. The clinician should document a three-generation pedigree (at a minimum) that ascertains ALS, FTD, other dementias, parkinsonism, psychiatric disorders, and suicide.9,60 The pedigree should be reviewed for evidence of dominant transmission because the majority of ALS genes are dominant. However, nonpenetrance, incomplete family information, misdiagnosis, false paternity, early death, undisclosed adoption, and other circumstances may obscure a clear pattern. For example, the presence of affected siblings is classically associated with recessive inheritance, but in ALS it may also be seen in dominant transmission with nonpenetrance, early death, or other issue obscuring a parent’s genetic status.
In ALS patients who do not have a history of ALS or FTD in a first- or second-degree relative, empiric data may be offered to estimate the chance that relatives would develop ALS. The lifetime risk of ALS in first-degree relatives appears to be 1–3% (toward the higher end of this range in male relatives and toward the lower end in female relatives). Risks for second-degree relatives are increased to a lesser degree, and risks for more distantly related family members do not appear to be increased.3,61 It is appropriate to discuss this risk information with families of probands of European descent with no close family history of ALS or FTD. Data on familial clustering of ALS in other populations is not readily available, and the different geographic distribution of the C9orf72 repeat expansion is one reason why European-derived empiric data may not be applicable to individuals of non-European descent.
The likelihood of a known, highly penetrant mutation in apparently sporadic cases is approximately 11%, with C9orf72 accounting for the majority of this risk.7 A notable exception to this is cases of combined ALS/FTD, which are highly associated with C9orf72 repeat expansions (at frequencies as high as 88%),21 even in the context of a negative family history.
For ALS patients who have a first- or second-degree relative affected with ALS or FTD, genetic risk assessment for family members should be informed by pedigree analysis and/or results of genetic testing. If dominant transmission is considered a possibility, then the 50% chance that children could inherit an ALS mutation should be discussed. The likelihood that a known, highly penetrant mutation is present is estimated at 60–70%.7 The majority of combined cases of ALS/FTD with a positive family history of either condition are caused by C9orf72 repeat expansions.21
Genetic testing options
Genetic testing options for ALS now include Sanger sequencing of individual genes, assays for the C9orf72 repeat expansion, sequential testing of multiple genes, next-generation sequencing panels (which are typically “bundled” with a C9orf72 assay), and whole-exome sequencing. Currently available tests, with estimated turnaround times and costs, are listed in Table 1 . The information displayed in this Table was gathered by surveying the major commercial laboratories offering ALS genetic testing at the time of drafting this article. For the most current information, the website or staff of the specific laboratory should be consulted at the time of testing.
There are several reasons to consider offering C9orf72 repeat expansion testing to all patients with ALS of European or partially European descent, regardless of family history. The overall 7% frequency of the C9orf72 repeat expansion in sALS,7 relatively low cost of the test, potential import of the test result, and the current and imminent state of C9orf72-targeted therapeutic trials may justify this approach. For fALS patients who test negative for C9orf72 expansion and for non-European patients with fALS, multigene panel testing should be offered (see Figure 2 for proposed testing schema). In our own clinic-based experience, this approach identifies a pathogenic mutation in 63.6% of fALS cases,62 which concurs with published literature.7 One potential drawback of this approach is that it does not allow for identification of a second deleterious mutation or variant in patients who test positive for the C9orf72 expansion. Pedigrees with more than one segregating ALS mutation or variant have been identified at greater than expected frequencies.63,64 In such cases where C9orf72 repeat testing is positive, as a first step, confirmation of a repeat expansion in each affected family member is particularly important.

Genetic testing options for patients with amyotrophic lateral sclerosis (ALS). Genetic testing for patients with ALS should be considered on a case-by-case basis. This chart demonstrates available options for initial and follow-up genetic testing. *Early onset may be defined as onset of symptoms before age 50 years. **The yield of clinical genetic testing in sALS patients of non-European descent is unknown. ***Commercial NGS panels for ALS are typically “bundled” with a C9orf72 assay. NGS, next-generation sequencing.
Consideration should also be given to offering testing to sALS patients with early onset of symptoms (younger than 50 years). Although extensive data on the yield of genetic testing in this population are not available, limited evidence suggests that patients with early-onset ALS, in the context of a negative family history, may harbor de novo mutations.45 If C9orf72 testing is negative in a patient with early onset of symptoms, then multigene panel testing may be offered.
Although whole-exome sequencing has been fruitful in identifying ALS genes in research,65 the yield of whole-exome sequencing in the clinical evaluation of the ALS patient is unknown. In fALS pedigrees, there is frequently only one affected family member living and available for testing, thus limiting opportunities for segregation analysis. However, exome sequencing may be considered in certain circumstances if C9orf72 repeat expansion and panel testing are negative. sALS probands with early onset and both parents available for testing/variant filtering may be appropriate candidates for exome sequencing. fALS cases in which two or more affected individuals are available for testing may also be considered candidates for exome sequencing.
Pretest counseling
Pretest genetic counseling should help patients anticipate the possible impact of genetic testing on themselves and their family members. Patients who are cognitively impaired should be accompanied by a legal guardian or health-care proxy.60 The heterogeneity and variable penetrance of ALS genes should be emphasized. Patients and families should be informed of the limitations of genetic testing, including: (i) a negative result does not exclude a genetic basis or contribution to the proband’s ALS; (ii) the test may be uninformative if a variant of uncertain significance is identified; and (iii) positive results do not consistently allow prediction of disease course. Families who are not ready to undergo genetic testing may consider DNA banking to permit future testing.
Presymptomatic genetic testing
Presymptomatic testing may be offered to adult first-degree relatives of ALS patients with established mutations. At-risk family members may have many reasons for seeking presymptomatic testing: to reduce uncertainty, to plan for the future, to make health or lifestyle choices, and to make decisions about family planning.66 Historically, published guidelines for presymptomatic genetic testing in ALS have been tailored after protocols for Huntington disease and Alzheimer disease,9,67,68 which include pretest genetic counseling, baseline neurologic and cognitive assessment, psychological evaluation, in-person disclosure, presence of support person, and posttest genetic counseling. More recently, Benatar et al. studied presymptomatic testing in ALS and made detailed recommendations for practice. These authors suggest that the testing process should involve at least two genetic counseling sessions (which may include predecision, pretest, and posttest counseling) and specify that phone counseling is acceptable. A minimum time interval of at least 1 week is suggested between initial genetic counseling and the decision to undergo testing to allow sufficient time for individuals to assimilate information and make an informed decision.69
Irrespective of the structure of presymptomatic genetic counseling, individuals should be assessed for psychosocial readiness to undergo testing. Testing should be deferred in persons with active psychiatric conditions, current substance abuse, suicidal tendencies, or those without social support systems.69 Motivations for seeking testing should be explored during genetic counseling and the limitations of testing should be emphasized. A positive test result does not resolve uncertainty regarding penetrance or clinical heterogeneity; age of onset, symptomatology, and even whether disease will actually occur cannot be predicted in most cases. Additionally, it should be discussed that no proven health, lifestyle, or medical interventions can reduce the risk of disease at this time. Pretest counseling should also address concerns related to genetic privacy. It should be emphasized that while state and federal laws prohibit genetic discrimination for health insurance, these do not encompass life, disability, or long-term care insurance60; persons seeking presymptomatic testing should be encouraged to consider arranging the latter prior to undergoing testing.
Concern exists regarding the perceived psychological risk to persons undergoing presymptomatic genetic testing. Limited evidence suggests that increased posttest distress among at-risk family members who undergo testing electively following genetic counseling is transient and not clinically significant.70 In a retrospective study of 135 persons undergoing presymptomatic testing for Huntington disease, Dufrasne et al. found that all participants expressed satisfaction regarding their decision to undergo testing, and none had a catastrophic reaction.71 However, psychological harms in presymptomatic genetic testing for ALS have not been studied, and the complexity of ALS genetics and penetrance may limit the applicability of data derived from studies of testing in other conditions.
Posttest counseling
Individuals receiving a positive test result should be given the information regarding the gene, specific mutation, and any established genotype–phenotype correlations. Family history should be reviewed in the context of the inheritance pattern of the mutation, and implications and risks for family members, including offspring, should be reviewed. The possibility of incomplete penetrance should be discussed. fALS families often develop theories about why the disease occurs; these should be explored and respectfully addressed. Clinicians should provide families with a copy of the genetic test result to facilitate accurate testing of relatives, as well as a brief summary letter outlining the results. Such letters often serve as a vehicle to inform family members of the genetic diagnosis72 and are particularly important in ALS because probands may die or become unable to communicate before results are received.
Technical challenges in ALS genetic testing
The accuracy of laboratory methodologies for C9orf72 clinical testing has been questioned. Although the Southern blot is considered the gold standard for detection and sizing of repeat expansion, this test is expensive and not widely available.73 Most laboratories use repeat-primed PCR (RP-PCR), which identifies alleles with >60 repeats but cannot specify the number of repeats.17 In some cases, amplicon length analysis is also used and allows determination of the exact number of repeats up to 30. Akimoto et al.73 performed an international blinded study in 14 laboratories with samples from 78 affected patients and found wide variance in reported genotypes as compared to the reference Southern blot result. They identified significant rates of false positives and negatives, and one specimen could not be genotyped at all in 9/14 laboratories. They concluded that a combination of amplicon length analysis and RP-PCR should be used for research purposes and that Southern blotting should be used for clinical testing.
A particular challenge with multigene panels, including ALS panels, is the high rate of variants of uncertain significance encountered. The variant of uncertain significance (VUS) rate—excluding cases where a VUS is reported in addition to a pathogenic change—with commercial multigene ALS panels is as high as 9% (J. Roach, personal communication). VUS interpretation is particularly difficult in ALS because affected family members are often not available for segregation analysis. Emerging data suggest that rare variant burden plays a role in the etiology of ALS30,63 but is not helpful in VUS interpretation in individual cases.
In considering the potential significance of a given VUS, it is helpful to consult population databases to assess the frequency of the variant in large, ethnically matched populations. Available databases include the Exome Aggregation Consortium (http://exac.broadinstitute.org/), the 1000 Genomes Project (http://browser.1000genomes.org), and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar). Although variants reported in population databases may be less likely to be pathogenic, the presence of a variant in a population database does not exclude pathogenicity. The ALS Data Browser (http://alsdb.org/) is a catalog of genetic variants identified from 1,424 Caucasian ALS patients that estimates the pathogenic potential of each variant in the form of Combined Annotation-Dependent Depletion scores. Variant assessment should also include review of the medical literature. If affected family members are available for testing, segregation analysis can be a powerful tool in variant interpretation. Despite these approaches, however, uncertainty regarding the significance of a VUS often remains. DNA banking is an option in these cases to enable testing of additional genes and/or segregation analysis in the future.
Summary
Current management guidelines for ALS do not address the issue of genetic testing.74 Recent progress in gene discovery and technology has both complicated and empowered the process of genetic risk assessment, genetic testing, and counseling in ALS. A relatively short time ago, genetic testing in ALS was limited to SOD1 sequencing; clinicians now have a wide array of testing options, including the C9orf72 repeat expansion assay, multigene NGS panels, and whole-exome sequencing. However, fundamental clinical skills such as the family history, pedigree analysis, and risk assessment remain critical to the proper application of genetic testing and counseling in this population. Genetic testing may help ALS patients understand the basis of their condition, allow accurate risk assessment and genetic testing of family members, and open the door for genotype-specific treatments. As the genetic basis of the remainder of fALS, and potentially sALS, is unraveled, genetic testing and counseling will become increasingly vital and should be incorporated into the routine management of ALS.
Disclosure
S.J.K. is supported by grant funding from the NINDS. The other authors declare no conflicts of interest.
References
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–591.
Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 2006;1762:956–972.
Gibson SB, Figueroa KP, Bromberg MB, Pulst SM, Cannon-Albright L. Familial clustering of ALS in a population-based resource. Neurology 2014;82:17–22.
Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology 1997;48:55–57.
Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G. Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5:208–212.
Cudkowicz ME, McKenna-Yasek D, Sapp PE, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol 1997;41:210–221.
Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17–23.
Agrawal M, Biswas A. Molecular diagnostics of neurodegenerative disorders. Front Mol Biosci 2015;2:54.
Chiò A, Battistini S, Calvo A, et al.; ITALSGEN Consortium. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 2014;85:478–485.
Talbot K. Should all patients with ALS have genetic testing? J Neurol Neurosurg Psychiatry 2014;85:475.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256.
Renton AE, Majounie E, Waite A, et al.; ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268.
Wagner K, Nagaraja H, Allain D, Quick A, Kolb SJ, Roggenbuck J. Genetic Testing in Amyotrophic Lateral Sclerosis: A Perspective from the ALS Community. Notheast ALS Consortium Annual Meeting, Tampa, FL, 5 November 2015.
Peters OM, Brown RH Jr . Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125:245–252.
Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014;10:661–670.
Majounie E, Renton AE, Mok K, et al.; Chromosome 9-ALS/FTD Consortium; French research network on FTLD/FTLD/ALS; ITALSGEN Consortium. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330.
Cruts M, van der Zee J, Van Broeckhoven C. C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia. In: Pagon RA, Ardinger HH, Wallace SE, et al. (eds). GeneReviews. University of Washington: Seattle, WA, 2015.
Cistaro A, Pagani M, Montuschi A, et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging 2014;41:844–852.
Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135(Pt 3):765–783.
Devenney E, Hornberger M, Irish M, et al. Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol 2014;71:331–339.
Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci 2013;36:450–459.
van der Zee J, Gijselinck I, Dillen L, et al.; European Early-Onset Dementia Consortium. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34:363–373.
Gijselinck I, Van Mossevelde S, van der Zee J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 2016;21:1112–1124.
Nordin A, Akimoto C, Wuolikainen A, et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet 2015;24:3133–3142.
Benussi L, Rossi G, Glionna M, et al. C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: a genotype-phenotype correlation study. J Alzheimers Dis 2014;38:799–808.
Scarrott JM, Herranz-Martín S, Alrafiah AR, Shaw PJ, Azzouz M. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin Biol Ther 2015;15:935–947.
Rosen DR, Patterson D, Figlewicz DA, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62.
Chiò A, Traynor BJ, Lombardo F, et al. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008;70:533–537.
Alavi A, Nafissi S, Rohani M, et al. Genetic analysis and SOD1 mutation screening in Iranian amyotrophic lateral sclerosis patients. Neurobiol Aging 2013;34:1516.e1–1516.e8.
Kenna KP, McLaughlin RL, Byrne S, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet 2013;50:776–783.
Millecamps S, Salachas F, Cazeneuve C, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet 2010;47:554–560.
Andersen PM, Forsgren L, Binzer M, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 1996;119 (Pt 4):1153–1172.
Själander A, Beckman G, Deng HX, Iqbal Z, Tainer JA, Siddique T. The D90A mutation results in a polymorphism of Cu,Zn superoxide dismutase that is prevalent in northern Sweden and Finland. Hum Mol Genet 1995;4:1105–1108.
Al-Chalabi A, Andersen PM, Chioza B, et al. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998;7:2045–2050.
Lopate G, Baloh RH, Al-Lozi MT, et al. Familial ALS with extreme phenotypic variability due to the I113T SOD1 mutation. Amyotroph Lateral Scler 2010;11:232–236.
Chiò A, Calvo A, Mazzini L, et al.; PARALS. Extensive genetics of ALS: a population-based study in Italy. Neurology 2012;79:1983–1989.
Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133.
Borroni B, Archetti S, Del Bo R, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res 2010;13:509–517.
Synofzik M, Born C, Rominger A, et al. Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol Aging 2014;35:1212.e1–1212.e5.
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208.
Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211.
Waibel S, Neumann M, Rosenbohm A, et al. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur J Neurol 2013;20:540–546.
Kinsley L, Siddique T. Amyotrophic lateral sclerosis overview. In: Pagon RA, Ardinger HH, Wallace SE, et al. (eds). GeneReviews. University of Washington: Seattle, WA, 2015.
Huey ED, Ferrari R, Moreno JH, et al. FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging 2012;33:1016.e9–1016.17.
Hübers A, Just W, Rosenbohm A, et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging 2015;36:3117.e1–3117.e6.
Johnson JO, Mandrioli J, Benatar M, et al.; ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–864.
Bersano A, Del Bo R, Lamperti C, et al. Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol Aging 2009;30:752–758.
Koppers M, van Blitterswijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:837.e7–837.13.
Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 2009;84:511–518.
Johnson JO, Pioro EP, Boehringer A, et al.; ITALSGEN Consortium. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 2014;17:664–666.
Leblond CS, Gan-Or Z, Spiegelman D, et al. Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2016;37:209.e17–209.e21.
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014;137(Pt 8):2329–2345.
Chaussenot A, Le Ber I, Ait-El-Mkadem S, et al.; French research network on FTD and FTD-ALS. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol Aging 2014;35:2884.e1–2884.e4.
Müller K, Andersen PM, Hübers A, et al. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014;137(Pt 12):e309.
Kurzwelly D, Krüger S, Biskup S, Heneka MT. A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain 2015;138(Pt 9):e376.
Ronchi D, Riboldi G, Del Bo R, et al. CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 2015;138(Pt 8):e372.
Chio A, Mora G, Sabatelli M, et al. CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 2015;36:1767 e1763–1766.
Zhang M, Xi Z, Zinman L, et al. Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 2015;138(Pt 9):e380.
Dols-Icardo O, Nebot I, Gorostidi A, et al.; Dementia Genetics Spanish Consortium (DEGESCO). Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 2015;138(Pt 12):e400.
Fong JC, Karydas AM, Goldman JS. Genetic counseling for FTD/ALS caused by the C9ORF72 hexanucleotide expansion. Alzheimers Res Ther 2012;4:27.
Hanby MF, Scott KM, Scotton W, et al. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain 2011;134(Pt 12):3454–3457.
Roggenbuck J, Kissel JT, Kolb SJ. Yield of Clinical Genetic Testing for ALS-Associated Mutations in a Tertiary Care ALS Clinic. National Society of Genetic Counselors: Pittsburgh, PA, 2015.
van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:3776–3784.
Cady J, Allred P, Bali T, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 2015;77:100–113.
Cirulli ET, Lasseigne BN, Petrovski S, et al.; FALS Sequencing Consortium. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015;347:1436–1441.
Williamson J, LaRusse S. Genetics and genetic counseling: recommendations for Alzheimer’s disease, frontotemporal dementia, and Creutzfeldt-Jakob disease. Curr Neurol Neurosci Rep 2004;4:351–357.
Goldman JS, Hahn SE, Catania JW, et al.; American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med 2011;13:597–605.
Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol 2012;19:360–375.
Benatar M, Stanislaw C, Reyes E, et al. Presymptomatic ALS genetic counseling and testing: Experience and recommendations. Neurology 2016;86:2295–2302.
Lerman C, Croyle RT, Tercyak KP, Hamann H. Genetic testing: psychological aspects and implications. J Consult Clin Psychol 2002;70:784–797.
Dufrasne S, Roy M, Galvez M, Rosenblatt DS. Experience over fifteen years with a protocol for predictive testing for Huntington disease. Mol Genet Metab 2011;102:494–504.
Baker DL, Eash T, Schuette JL, Uhlmann WR. Guidelines for Writing Letters to Patients. J Genet Couns 2002;11:399–418.
Akimoto C, Volk AE, van Blitterswijk M, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 2014;51:419–424.
Miller RG, Jackson CE, Kasarskis EJ, et al.; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73:1227–1233.
Acknowledgements
S.J.K. is supported by grant funding from the NINDS (K08NS067282 and U01NS079163). The ALS/MND Multidisciplinary Clinic and Research Program at the Ohio State University Wexner Medical Center is grateful for the Julie Bonasera ALS Fund and the Fred F. and Herman M. Dreier ALS Fund for continued support of our Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Roggenbuck, J., Quick, A. & Kolb, S. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 19, 267–274 (2017). https://doi.org/10.1038/gim.2016.107
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.107
Keywords
This article is cited by
-
Targeting Sigma Receptors for the Treatment of Neurodegenerative and Neurodevelopmental Disorders
CNS Drugs (2023)
-
Anti-apoptotic Splicing Variant of AIMP2 Recover Mutant SOD1-Induced Neuronal Cell Death
Molecular Neurobiology (2023)
-
Targeted sequencing panels in Italian ALS patients support different etiologies in the ALS/FTD continuum
Journal of Neurology (2021)
-
Exploring neurologists’ perspectives on the return of next generation sequencing results to their patients: a needed step in the development of guidelines
BMC Medical Ethics (2018)
-
Glycoprotein NMB: an Emerging Role in Neurodegenerative Disease
Molecular Neurobiology (2018)
