
Abstract
Purpose:
The genetic etiology of atrioventricular septal defect (AVSD) is unknown in 40% cases. Conventional sequencing and arrays have identified the etiology in only a minority of nonsyndromic individuals with AVSD.
Methods:
Whole-exome sequencing was performed in 81 unrelated probands with AVSD to identify potentially causal variants in a comprehensive set of 112 genes with strong biological relevance to AVSD.
Results:
A significant enrichment of rare and rare damaging variants was identified in the gene set, compared with controls (odds ratio (OR): 1.52; 95% confidence interval (CI): 1.35–1.71; P = 4.8 × 10−11). The enrichment was specific to AVSD probands, compared with a cohort without AVSD with tetralogy of Fallot (OR: 2.25; 95% CI: 1.84–2.76; P = 2.2 × 10−16). Six genes (NIPBL, CHD7, CEP152, BMPR1a, ZFPM2, and MDM4) were enriched for rare variants in AVSD compared with controls, including three syndrome-associated genes (NIPBL, CHD7, and CEP152). The findings were confirmed in a replication cohort of 81 AVSD probands.
Conclusion:
Mutations in genes with strong biological relevance to AVSD, including syndrome-associated genes, can contribute to AVSD, even in those with isolated heart disease. The identification of a gene set associated with AVSD will facilitate targeted genetic screening in this cohort.
Genet Med 18 2, 189–198.
Similar content being viewed by others

Introduction
Atrioventricular septal defect (AVSD) is a relatively rare disorder, representing 7% of all congenital heart defects (CHDs).1 Although AVSD is most often associated with trisomy 21 or heterotaxy, ~40% of patients have neither of these associations. Within this etiologically diverse group of patients, ~30% have an identifiable genetic diagnosis relating to a chromosomal or single-gene disorder, 10% have nondiagnostic dysmorphic features or extracardiac anomalies, and the remainder have no discernible extracardiac anomalies (nonsyndromic AVSD).1,2 To date, targeted sequencing3,4,5 and interrogation for large copy-number variants6 have identified the etiology in only a minority of nonsyndromic individuals with AVSD. Traditional gene discovery strategies are limited in AVSD as a result of locus heterogeneity, possible reduced penetrance, and relatively small numbers of cases and families. We recently reported results of an unsupervised analysis of whole-exome sequencing (WES) data in AVSD probands that identified rare causal variants in NR2F2.7 To identify additional genetic causes in this cohort, we analyzed potentially causal variants in all known genes with biological relevance to AVSD.
Materials and Methods
Study population
Patients were prospectively enrolled in an Ontario province-wide Heart Centre Biobank registry after informed consent. The study was approved by the research ethics boards of the Hospital for Sick Children and all other participating sites.8,9 The primary cohort included unrelated Caucasian probands with AVSD. Principal component analysis was performed to confirm overlap with individuals of Caucasian ancestry included in HapMap (Supplementary Figure S1 online). Patients with an identified chromosomal or syndromic disorder or a situs anomaly were excluded. Detailed cardiac and extracardiac features were assessed through review of medical records, imaging, and dysmorphology assessment when possible.
Data for replication analysis (n = 81) were obtained from WES of 44 AVSD probands (GO-CHD, Oxford registry; Leuven, Belgium) provided by the Wellcome Trust Sanger Institute (previously described)7 and 37 AVSD probands from the CONCOR registry DNA bank of the University Cardiology Institute Netherlands and the Ontario Heart Centre Biobank Registry who underwent targeted sequencing. All probands in the replication cohort were unrelated, nonsyndromic, and Caucasian. Sixty-four unrelated Caucasian probands from the Heart Centre Biobank Registry with isolated tetralogy of Fallot (TOF) underwent WES as a non-AVSD CHD cohort.
Variant calls from WES of 4,300 individuals of European American ancestry from the National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server10 (EVS) served as controls (“EVS controls”). Additional controls included whole-genome sequencing data from 40 unaffected parents of children with autism spectrum disorders (S. Walker and S.W. Scherer, personal communication), and targeted genotyping in 97 unaffected, Caucasian, population-matched controls without heart disease from Ontario.
Whole-exome sequencing
WES was performed using the Agilent SureSelect Target Enrichment Kit (V3 50 Mb; Agilent Technologies, Santa Clara, CA) for sequence capture and Illumina HiSeq2500 (75-bp paired-end reads) for sequencing (Illumina, San Diego, CA) to a target depth of 100×, as previously described.7 The non-AVSD cohort was sequenced using the Agilent SureSelect Target Enrichment Kit (V4 51 Mb; Agilent Technologies) for sequence capture and Illumina HiSeq (90-bp paired-end reads) for sequencing (Illumina) (BGI Genomics, Philadelphia, PA) to a target depth of 50×.
AVSD gene list
The literature was systematically reviewed to curate all genes associated with AVSD identified in previous human and animal studies (Supplementary Tables S1 and S1.2 online). This generated a list of 112 AVSD genes, including 47 associated with human syndromes, 30 with animal models, 20 with both human and animal references, 9 with isolated AVSD in humans, and 6 reported as cardiac modifiers in trisomy 21. Of these 112 genes, 58 were associated with syndromic CHD (“syndromic” genes).
Variant filtering
Variants in these 112 genes were filtered for quality, leaving only calls with a passing Genome Access Toolkit (software) score (99% accuracy in read determination)11 and read-depth coverage of ≥30. Nonsynonymous, nonsynonymous splice, stop-gain, and stop-loss variants were prioritized. The variants were annotated with Annovar12 to obtain the minor allele frequency for each variant from the 1000 Genomes Project13 and EVS.10 Rare variants, defined as those with a minor allele frequency <0.01 in the 1000 Genomes Project and EVS European American ancestry, were prioritized and included novel variants, that is, variants not present in either the 1000 Genomes Project, EVS data sets, or in the Database of Single Nucleotide Polymorphisms.14 Variants were annotated with SIFT15 and PolyPhen2,16 and any variant predicted to be pathogenic in at least one program was called damaging. Evolutionary conservation was defined as PhyloP score >0 (range: −14 to +6; sites predicted to be conserved are assigned positive scores)17 and/or PhastCONS ≥0.8 (range: 0–1, where 1 indicates a highly conserved locus).18
Mutation burden analysis
The overall burden of rare (including novel) nonsynonymous variants and rare synonymous variants in the AVSD, EVS, and non-AVSD CHD cohorts was assessed across all 112 genes. The EVS does not provide sample-level information; therefore, for consistency, the burden analysis assumes that each genotype represents an independent sample. Variant data from EVS control and non-AVSD CHD cohorts were filtered in the same manner as the case data. To assess mutation burden, Fisher’s exact test was performed across all 112 genes for the number of individuals with and without rare and rare damaging nonsynonymous and rare synonymous variants among cases and controls. A control burden analysis also was performed using a list of 11 autism spectrum disorder genes19 (refer to Supplementary Figure S2 online).
Coverage analysis
To ensure that a difference in mutation burden across these 112 genes was not secondary to differences in the coverage of genomic regions between cohorts, coverage analysis was performed among cases and controls. Consensus coding sequence exon coordinates for all exons in 112 genes were extracted from University of California Santa Cruz Table Browser,20 and the mean depth per coding base pair was calculated across all BAM files, as previously described. The percentage of bases covered to a depth of ≥20 for 112 genes was calculated and the distribution was compared between AVSD, EVS, and non-AVSD CHD cohorts using the Student’s t-test (two-tailed). A nucleotide-by-nucleotide coverage analysis then was undertaken to verify adequate case and control coverage at each locus corresponding to variant location. Variants were excluded from the mutation burden calculation if the average depth of coverage in either cohort was <20.
Variant validation
All prioritized variants were verified by Sanger’s dideoxynucleotide sequencing. The amplified products were subjected to dideoxynucleotide polymerase chain reaction amplification and were sequenced on 3730xl DNA Analyzer (Applied Biosystems/Life Technologies, Grand Island, NY; Supplementary Table S2.1 online). The sequence chromatograms were aligned to primer target regions using CodonCode Aligner (CodonCode, Centerville, MA). Parental genotypes were evaluated if parental DNA was available.
Replication
Genes enriched for rare (including novel) and rare damaging nonsynonymous variants in AVSD cases versus EVS controls on mutation burden analysis were selected for targeted sequencing in the replication cohort using Agilent Haloplex Custom Target Enrichment (Agilent Technologies). A custom probe set was designed using Agilent SureDesign web-based software to target all coding exons for the prioritized genes (http://earray.chem.agilent.com/suredesign). Sequencing (100-bp paired-end reads, targeted at >200× coverage) was performed using Illumina HiSeq2500 (Illumina) at The Centre for Applied Genomics in The Hospital for Sick Children. Agilent SureCall software was used to generate BAM files and call variants. Variants were annotated and filtered using the same protocol used for the primary cohort. To assess the population frequency of the recurring variant in MDM4 (8:204518457 A>C (K324Q, rs41299595)), genotyping was performed in 97 Caucasian controls without heart disease from Ontario using a custom-designed TaqMan SNP Assay (Life Technologies; Supplementary Table S2.2 online). Samples were analyzed using the ViiA 7 Real-Time PCR System with ViiA7 software (Applied Biosystems, Life Technologies, Carlsbad, CA).
Results
Of 260 individuals with AVSD enrolled in the Heart Centre Biobank Registry, 83 probands were eligible for inclusion and underwent WES ( Table 1 ). Variants in 112 genes from 81 unrelated probands (2 probands did not sequence) were filtered to identify 163 unique rare variants, of which 91 were predicted to be damaging (Supplementary Figure S3 online).
The AVSD cohort was enriched for rare and rare damaging variants
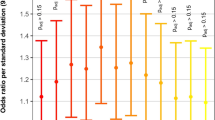
A mutation burden analysis across all 112 genes was conducted to assess for enrichment of rare variants in the AVSD cohort compared with EVS controls. There was no difference between the two cohorts in the frequency of rare synonymous variants (1.6 vs. 1.6%; odds ratio (OR): 1.01; P = 0.9). The frequency of rare nonsynonymous variants, however, was higher in the AVSD cohort (3.2%) compared with the EVS controls (2.1%; OR: 1.52; P = 4.8 × 10−11), as was the frequency of rare damaging variants in the AVSD cohort (1.5%) versus EVS controls (1.2%; OR: 1.26; P = 0.01; Figure 1 and Supplementary Table S3.1 online). We also observed an enrichment of rare nonsynonymous variants in the AVSD cohort versus EVS controls when comparing only the subset of 58 syndromic genes (OR: 1.32; P = 0.001) (Supplementary Table S3.1 online). There was no enrichment of rare or rare damaging nonsynonymous variants in the AVSD cohort compared with the EVS cohort when the same analysis was conducted with a control set of 11 autism spectrum genes (Supplementary Figure S2 online).

Mutation burden analysis across 112 genes with biological relevance to atrioventricular septal defect (AVSD). All rare, nonsynonymous variants in the gene were collapsed for each individual to allow a patient-level analysis. Odds ratio (OR) for the number of individuals with rare nonsynonymous, rare damaging nonsynonymous, and synonymous variants across 112 genes in the AVSD cohort compared with controls from the Exome Variant Server (EVS) and non-AVSD congenital heart defect (CHD) cohort with P values are shown. The AVSD cohort showed enrichment of rare nonsynonymous variants (OR: 1.5; P = 4.7 × 10−11) and rare damaging nonsynonymous variants (OR: 1.3; P = 0.01) compared with the EVS cohort. The AVSD cohort showed a similar enrichment of rare nonsynonymous variants (OR: 2.3; P = 2.2 × 10−14) and rare damaging nonsynonymous variants (OR: 2.1; P = 2.1 × 10−6) compared with the non-AVSD CHD cohort. There was no difference in rare synonymous variants between the cohorts (AVSD versus EVS, OR: 1.0; P = 0.9 and AVSD versus non-AVSD CHD, OR: 1.2; P = 0.2).
To assess whether the enrichment was specific to AVSD or also occurred in other forms of CHD, mutation burden was compared with that of a non-AVSD CHD cohort (TOF). Again, there was no significant difference between the cohorts in the frequency of rare synonymous variants (1.6 vs. 1.4%; OR: 1.12; P = 0.3; Figure 1 and Supplementary Table S3.2 online). However, the AVSD cohort was enriched for rare nonsynonymous variants in the 112 genes (3.2 vs. 1.4%; OR: 2.33; P = 2.2 × 10−14) and rare damaging nonsynonymous variants (1.5 vs. 0.7%; OR: 2.11; P = 2.1 × 10−6) compared with the non-AVSD CHD cohort. Enrichment of variants in the AVSD versus non-AVSD CHD cohort also was seen when analysis was limited to only 58 syndromic genes (Supplementary Table S3.2 online).
To ensure that enrichment of variants in the AVSD cohort was not related to differences in coverage between cohorts, we compared coverage across the 112 genes in the AVSD cohort versus the EVS control and non-AVSD CHD cohorts. The mean percentage of bases covered to a depth of ≥20 for all 112 genes was similar between the AVSD (83.0 ± 22%) and the non-AVSD CHD cohort (81.8 ± 14.6%; P = 0.63) but lower than that among the EVS controls (89.7 ± 18.9%; P = 0.02). Therefore the higher mutation burden in AVSD cases was not due to underreporting of variants in controls or in the non-AVSD CHD cohort (Supplementary Figure S4 online). The enrichment for rare and rare damaging variants in AVSD cases remained significant when analysis was corrected for nucleotide-by-nucleotide coverage by excluding variants that did not meet the depth cutoffs in both cohorts (Supplementary Tables S3.1 and S3.2 online).
Genotype–phenotype associations
As reported previously, NR2F2 was significantly enriched for both rare and rare damaging variants.7 Other than NR2F2, six genes showed significant enrichment for rare and novel nonsynonymous variants; MDM4 remained significant after correcting for multiple testing across 112 genes. These genes—NIPBL, CHD7, CEP152, BMPR1a, ZFPM2, and MDM4—were prioritized for validation by Sanger sequencing, replication, and determination of inheritance patterns ( Table 2 and Supplementary Figure S5 online). Across the six prioritized genes, 23 probands had rare/novel damaging variants ( Tables 3 and 4 and Supplementary Table S4 online). Given that many of these genes are syndrome-associated, patients were assessed further for extracardiac anomalies. Genotype–phenotype associations for individual genes are described below.
NIPBL. Six AVSD probands (7.4%) had rare nonsynonymous variants in NIPBL compared with 2.3% in EVS controls (OR: 3.3; P = 0.02; Table 2 ). The M1318V and S2471T variants were novel, highly conserved, and predicted to be damaging ( Table 3 and Supplementary Table S4 online). The N105D and N393K variants each previously occurred in a single individual among 4,300 EVS European American controls and not in the other control data sets. Both variants are conserved, and the N105D variant is predicted to be damaging. The A179T variant (AVSD_65) was also identified in one individual in the replication cohort (Supplementary Table S5 online) and in one non-AVSD CHD proband. Although it was previously reported in 29 individuals in the European American EVS, and in the 1000 Genomes Project and the Database of Single Nucleotide Polymorphisms, it is listed in the Human Gene Mutation Database as a potentially disease-causing variant (Supplementary Table S4 online). An additional variant, I314V, was identified in the replication cohort (Supplementary Table S5 online) and has not been previously reported in public data sets. NIPBL mutations are known to be associated with Cornelia de Lange syndrome (CDL). None of the AVSD probands with NIPBL variants had clinical characteristics of CDL ( Table 4 ), however, although two probands had associated semilunar valve anomalies (AVSD_33 and AVSD_72), which are commonly associated with CDL.
CHD7. Nine AVSD probands (11.1%) had rare nonsynonymous variants in CHD7 compared with 5.0% of EVS controls (OR = 2.3; P = 0.04; Table 2 ). The T93A, Y1325H, A2105V, and S2383C variants were all novel or exceptionally rare (Y1325H occurred in one EVS individual). All were conserved, and the A2105V, Y1325H, and S2383C variants were predicted to be damaging. The M340V and M2527L variants occurred in probands and in the replication cohort (Supplementary Table S5 online) and also were seen in the EVS data set (62 and 43 individuals); these were predicted to be benign. Three additional rare nonsynonymous variants were identified in the replication cohort: A1950T, P2083S, and L935F (Supplementary Table S5 online). None of the variants occurred in 40 unaffected parents from an Ontario-based cohort of families with autism spectrum disorders. CHD7 mutations are known to be associated with CHARGE (Coloboma, Heart, Choanal Atresia, Retardation, Genital and Ear Anomalies) syndrome. However, six of nine probands with CHD7 variants had isolated cardiac disease. One proband had dysmorphic features and extracardiac anomalies but none of the diagnostic features of CHARGE syndrome. The other two probands had single minor anomalies unrelated to CHARGE syndrome ( Table 4 ).
CEP152. Rare nonsynonymous variants in CEP152 occurred in 9.7% of AVSD cases compared with 4.3% of EVS controls (OR: 2.4; P = 0.03; Table 2 ). The L581I variant was novel, whereas the T1524A, L1105V, W960R, I294V, and S85R variants all were rare. Only the W960R variant was highly conserved and predicted to be damaging. Two additional variants were identified in the replication cohort: G181D and R115Q (Supplementary Table S5 online). None of the variants occurred in 40 unaffected parents from an Ontario-based cohort of families with autism spectrum disorders. All variants were heterozygous, and none of the probands had features consistent with Seckel syndrome, dwarfism, or microcephaly, which are known to be associated with defects in CEP152 ( Table 4 ).
BMPR1a. Rare nonsynonymous variants in BMPR1a were identified in 3.7% of AVSD cases compared with 0.7% of EVS controls (OR: 5.3; P = 0.02; Table 2 ). All three variants (R478H, D429V, and P481S) were exceptionally rare and predicted to be damaging ( Table 4 ). Two probands (AVSD_2 and AVSD_17) had isolated cardiac disease, and AVSD_57 also had learning and psychiatric disabilities and cervical spine anomalies. AVSD_17 and AVSD_57 both had a left superior vena cava to coronary sinus ( Table 4 ).
ZFPM2. Rare nonsynonymous variants in ZFPM2 occurred in 13.6% of AVSD cases compared with 6.7% of EVS controls (OR: 2.2; P = 0.03). Two variants (P361S and G885S) were exceptionally rare in EVS and absent in the other control data sets. Both these variants were conserved, and the P361S variant was predicted to be damaging. Six of the nine AVSD probands with ZFPM2 variants had a second variant in one of the six priority genes, including two probands with two heterozygous ZFPM2 variants. Of these, AVSD_53 had associated anomalies including pulmonary atresia with major aortopulmonary collaterals, as well as multiple extracardiac anomalies including a diaphragmatic hernia (P361S and E30G). The E30G, D98N, M544I, and V631I variants in the AVSD cohort and the S210T variant in the non-AVSD TOF cohort also were identified in the replication cohort, along with D1051Y, a novel variant. In addition, five individuals in the non-AVSD CHD cohort had ZFPM2 variants, none of which recurred in the AVSD cohort.
MDM4. Rare nonsynonymous variants in MDM4 occurred in 7.4% of AVSD cases compared with 1.2% of EVS controls (OR: 5.0; P = 3.2 × 10−4). One variant (K324Q) occurred in five individuals (minor allele frequency = 0.061), in one unaffected autism control (S. Walker and S.W. Scherer, personal communication), and in 40 EVS individuals (minor allele frequency = 0.0047). Because this single variant occurred with a high frequency in our cohort, genotyping was undertaken in 97 unaffected controls. The K324Q variant was found at a lower frequency in controls (2/97 or 2.1%) compared with AVSD cases (6.1%; P = 0.003). The P338A variant identified in AVSD_29 occurred in four EVS individuals but not in those in the 1000 Genomes Project or in unaffected autism controls. An additional three variants were identified in the replication cohort (Supplementary Table S5 online). All the probands with MDM4 variants had isolated cardiac disease.
Compound mutations
Of the 34 probands with variants in the six prioritized genes, eight probands had more than one rare or rare damaging nonsynonymous variant in more than one gene, of which six had a second mutation in ZFPM2, as detailed above. In addition, AVSD_15 had variants in CHD7, NIPBL, and CEP152 and AVSD_72 had variants in CEP152 and NIPBL. Interestingly, AVSD_15 and AVSD_72 shared the same NIPBL variant (N393K), which occurred in only one individual in the EVS.
Discussion
We conducted a comprehensive gene analysis using next-generation sequencing in a cohort of unrelated AVSD probands to identify genomic variants associated with AVSD. The strength of our approach included a comprehensive gene selection methodology identifying genes with biological associations to AVSD, the majority of which have not been systematically examined in a large cohort of patients with AVSD. There are three important findings from our study.
First, genes with known biological associations to AVSD were enriched for rare and rare damaging nonsynonymous variants compared with healthy controls. Mutation burden analysis at the gene level suggested an association between six genes (NIPBL, CHD7, CEP152, BMPR1a, ZFPM2, and MDM4) and AVSD, of which the association with MDM4 remained significant after multiple testing. We used stringent methods to ensure that the results remained true when analysis was adjusted for gene-by-gene and nucleotide-by-nucleotide coverage between cases and controls.
Second, we demonstrated enrichment of variants in 112 genes not only in AVSD cases compared with EVS controls but also compared with a non-AVSD cohort of TOF probands. This enrichment suggests that at least some of the associations were specific to AVSD and not to CHD in general and that there may be gene-specific cardiac phenotypes related to unique genetic contributions to cardiac morphogenesis.21,22 While a recent paper analyzed all CHD phenotypes together to identify de novo mutations,23 our results suggest the importance of studying probands with distinct cardiac phenotypes to improve the specificity of the observed associations and improve the predictive ability for clinical applications such as prenatal and reproductive counseling in the future.
Finally, “syndromic” genes were enriched for mutations despite the absence of classic syndromic features. These mutations occurred in patients with either isolated cardiac disease or subtle manifestations that did not meet diagnostic criteria for a clinical syndrome. In particular, three of the top-ranked associated genes were syndromic genes, that is, NIPBL, CHD7, and CEP152, suggesting that a search for mutations should include syndromic genes regardless of clinical phenotype.
An important syndromic gene was NIPBL, a causal gene for autosomal dominant CDL (OMIM 122470), a developmental disorder. Approximately 50% of patients with CDL have NIPBL variants, typically with milder phenotypes in those with missense mutations.24,25 Of patients with CDL, 30% have CHD, including septal defects, semilunar valve anomalies, or AVSD.1,26 To our knowledge, isolated CHD has not previously been reported with NIPBL mutations.27 In our cohort NIPBL was enriched for rare and rare damaging variants. Yet none of the probands had features of CDL syndrome ( Table 4 ), although we cannot rule out subtle manifestations because we were unable to recontact all the probands for dysmorphology assessment. These findings suggest a contributory role for NIPBL not only in CHD associated with CDL syndrome but also in cases with isolated AVSD.
Another syndromic gene enriched for variants was CHD7, the only gene known to cause CHARGE syndrome (OMIM 214800), a rare, autosomal dominant condition with a prevalence of up to 1:8,500 (ref. 29). This was not unique to AVSD but also occurred in the cohort with TOF, a lesion type known to be associated with CHARGE syndrome. Approximately 75% of individuals with CHARGE syndrome have CHD.28 Mutations in CHD7 are identified in over 90% of patients with the CHARGE phenotype, and incomplete penetrance has not previously been reported.29 CHD7 also was recently reported as part of the H3K4me gene mutations associated with CHD.23 In our study none of the nine individuals with CHD7 mutations had features of CHARGE syndrome.
CEP152, another syndromic gene, is involved in regulation of cellular response to DNA damage in Seckel syndrome.30 Seckel syndrome (OMIM 210600) is a rare, autosomal recessive disorder characterized by severe growth retardation, microcephaly, and mental retardation, as well as partial AVSD and other forms of CHD.31,32 CEP152 was enriched for rare nonsynonymous heterozygous variants in AVSD probands, none of whom had features of Seckel syndrome. While a causal role for CEP152 seems less compelling at a variant level, its role as a phenotype modifier cannot be excluded. Together, these findings reiterate that the absence of obvious syndromic features should not preclude a search for mutations in syndromic genes.
Three patients had three exceptionally rare or novel variants in BMPR1a ( Table 3 and Supplementary Table S4 online) that were highly conserved and predicted to be damaging. One proband (AVSD_17) had unbalanced AVSD with coarctation, the same cardiac phenotype as previously described in a patient with a 10q22q23 deletion encompassing BMPR1a.33 Bmpr1a (Alk3) knockout mice show embryonic lethality between embryonic days 10.5 and 11.5, with failure of endocardial cushion formation and atrioventricular valve and adjacent septal formation, consistent with a specific and critical role in the formation of the atrioventricular canal.21,22 Our findings are consistent with prior human and animal studies that implicate BMPR1a in the development of isolated AVSD.
ZFPM2 is a zinc-finger nuclear protein expressed in the developing heart and known to alter GATA4 transcriptional activity.34,35 Knockout mouse models demonstrate cardiac defects including AVSD,35,36 double-outlet right ventricle, TOF, and coronary artery anomalies. ZFPM2 variants have also been reported in patients with conotruncal defects, but not with AVSD.37,38 The enrichment of ZFPM2 variants in nine probands in our primary AVSD cohort and eight probands in the replication cohort is a new finding. Nonetheless, whether ZFPM2 variants play a causal or a modifier role in AVSD is unclear since the variants frequently cosegregated with variants in other candidate genes (Supplementary Table S4 online) and, although rare, occurred in multiple EVS cases. Also, the E30G variant was not specific to AVSD and has not been associated with functional effects on GATA4-mediated transcription and is therefore likely to be a benign polymorphism.39 This is in contrast to the majority (four of five) of the variants identified in the non-AVSD TOF cohort, which were novel (Supplementary Table S4 online).
Six AVSD probands had variants in MDM4, a p53 inhibitor, with MDM4 showing the strongest association with AVSD compared with the other five genes. MDM4 is involved in regulating endocardial epithelial-to-mesenchymal transition and subsequent proliferation of mesenchyme in the endocardial cushions. A mouse model heterozygous for Mdm2 and Mdm4 had impaired atrioventricular valvuloseptal development with significantly reduced mesenchymal cell density at the endocardial cushions.40 Two rare damaging, and conserved variants were identified, one of which was a recurring variant (K324Q) seen in five probands. The variant frequency was higher than that among controls, suggesting that this variant may potentially cause disease in AVSD cases.
There were some limitations to our study. Functional validation of individual variants was beyond the scope of this study; however, all variants were in genes with known association with CHD. We did not assess for structural variation, frameshift variants, or noncoding regulatory variants. A paucity of parental samples limited our ability to ascertain transmission of the variants or perform segregation analysis. In addition, local control data were limited; our control population was derived from public data sets, and therefore we corrected for two limitations: coverage and population stratification.
In summary, a biology-guided approach allowed us to identify enrichment of variants in genes that play a central role in cardiac development in general and in atrioventricular canal development in particular. There is compelling data to support a contributory role for NIPBL, CHD7, BMPR1a, and MDM4 gene variants, whereas the role of variants in ZFPM2 and CEP152 requires further investigation. The enrichment of variants in “syndromic” genes highlights the importance of not excluding these genes when investigating patients with nonsyndromic phenotypes. A targeted search for variation in lesion-specific genes within next-generation sequencing data has the potential to reduce time and cost of bioinformatics analysis and to increase the specificity and predictive accuracy of the observed associations.
Disclosure
The authors declare no conflict of interest.
References
Ferencz C, Loffredo CA, Correa-Villasenor A, Wilson PD. Atrioventricular septal defects with and without Down syndrome. In: Ferencz C, Loffredo CA, Correa-Villasenor A, Wilson PD (eds). Genetic & Environmental Risk Factors of Major Cardiovascular Malformations: the Baltimore-Washington Infant Study 1981–1989, 1997. Futura Publishing: Armonk, 1997:103–122.
Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Atrioventricular canal defect without Down syndrome: a heterogeneous malformation. Am J Med Genet 1999;85:140–146.
Robinson SW, Morris CD, Goldmuntz E, et al. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet 2003;72:1047–1052.
Sarkozy A, Lepri F, Marino B, Pizzuti A, Digilio MC, Dallapiccola B. Additional evidence that PTPN11 mutations play only a minor role in the pathogenesis of non-syndromic atrioventricular canal defect. Am J Med Genet A 2006;140:1970–1972.
Smith KA, Joziasse IC, Chocron S, et al. Dominant-negative ALK2 allele associates with congenital heart defects. Circulation 2009;119:3062–3069.
Priest JR, Girirajan S, Vu TH, Olson A, Eichler EE, Portman MA. Rare copy number variants in isolated sporadic and syndromic atrioventricular septal defects. Am J Med Genet A 2012;158A:1279–1284.
Al Turki S, Manickaraj AK, Mercer CL, et al.; UK10K Consortium. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet 2014;94:574–585.
Papaz T, Safi M, Manickaraj AK, et al. Factors influencing participation in a population-based biorepository for childhood heart disease. Pediatrics 2012;130:e1198–e1205.
Fung A, Manlhiot C, Naik S, et al. Impact of prenatal risk factors on congenital heart disease in the current era. J Am Heart Assoc 2013;2:e000064.
Exome Variant Server, NHLBI Grand Opportunity Exome Sequencing Project (ESP) Seattle, WA. http://evs.gs.washington.edu/EVS. Accessed 22 August 2013.
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
The 1000 Genomes Project Consortium. A map of human genome variation from population scale sequencing. Nature 2010;467:1016–1073.
Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–121.
Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034–1050.
Yuen RKC, Thirubahindrapuram B, Merico D, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 2015;21:185–191.
Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004;32(Database issue):D493–D496.
Park C, Lavine K, Mishina Y, Deng CX, Ornitz DM, Choi K. Bone morphogenetic protein receptor 1A signaling is dispensable for hematopoietic development but essential for vessel and atrioventricular endocardial cushion formation. Development 2006;133:3473–3484.
Kaneko K, Li X, Zhang X, Lamberti JJ, Jamieson SW, Thistlethwaite PA. Endothelial expression of bone morphogenetic protein receptor type 1a is required for atrioventricular valve formation. Ann Thorac Surg 2008;85:2090–2098.
Zaidi S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013;498:220–223.
Gillis LA, McCallum J, Kaur M, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet 2004;75:610–623.
Selicorni A, Colli AM, Passarini A, et al. Analysis of congenital heart defects in 87 consecutive patients with Brachmann-de Lange syndrome. Am J Med Genet A 2009;149A:1268–1272.
Chatfield KC, Schrier SA, Li J, et al. Congenital heart disease in Cornelia de Lange syndrome: phenotype and genotype analysis. Am J Med Genet A 2012;158A:2499–2505.
Deardorff MA, Clark DM, Krantz ID. Cornelia de Lange syndrome. GeneReviews, 2005 (updated 27 October 2011). http://www.ncbi.nlm.nih.gov/books/NBK1104/.
Corsten-Janssen N, Kerstjens-Frederikse WS, du Marchie Sarvaas GJ, et al. The cardiac phenotype in patients with a CHD7 mutation. Circ Cardiovasc Genet 2013;6:248–254.
Lalani SR, Hefner MA, Belmont JW, Davenport SLH. CHARGE syndrome. In: GeneReviews, 2006 (updated 2 February 2012). http://www.ncbi.nlm.nih.gov/books/NBK1117/.
Kalay E, Yigit G, Aslan Y, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 2011;43:23–26.
Ucar B, Kilic Z, Dinleyici EC, Yakut A, Dogruel N. Seckel syndrome associated with atrioventricular canal defect: a case report. Clin Dysmorphol 2004;13:53–55.
Rappen U, von Brenndorff AI. [Cardiac symptoms in 2 patients with Seckel syndrome]. Monatsschr Kinderheilkd 1993;141:584–586.
Breckpot J, Tranchevent LC, Thienpont B, et al. BMPR1A is a candidate gene for congenital heart defects associated with the recurrent 10q22q23 deletion syndrome. Eur J Med Genet 2012;55:12–16.
Svensson EC, Tufts RL, Polk CE, Leiden JM. Molecular cloning of FOG-2: a modulator of transcription factor GATA-4 in cardiomyocytes. Proc Natl Acad Sci USA 1999;96:956–961.
Tevosian SG, Deconinck AE, Tanaka M, et al. FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell 2000;101:729–739.
Katz SG, Williams A, Yang J, et al. Endothelial lineage-mediated loss of the GATA cofactor Friend of GATA 1 impairs cardiac development. Proc Natl Acad Sci USA 2003;100:14030–14035.
Huang X, Niu W, Zhang Z, et al. Identification of novel significant variants of ZFPM2/FOG2 in non-syndromic Tetralogy of Fallot and double outlet right ventricle in a Chinese Han population. Mol Biol Rep 2014;41:2671–2677.
De Luca A, Sarkozy A, Ferese R, et al. New mutations in ZFPM2/FOG2 gene in tetralogy of Fallot and double outlet right ventricle. Clin Genet 2011;80:184–190.
Pizzuti A, Sarkozy A, Newton AL, et al. Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of Fallot. Hum Mutat 2003;22:372–377.
Zhang Q, He X, Chen L, et al. Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J Pathol 2012;228:416–428.
Acknowledgements
The authors acknowledge the SickKids Labatt Family Heart Centre Biobank Registry for DNA samples; Mina Safi, Tanya Papaz, and Carly Ogaki for patient recruitment and follow-up; Elizabeth Uleryk, director of the SickKids Hospital Library and Archives, for assistance with systematic literature review; The SickKids Centre for Applied Genomics for targeted resequencing and genotyping; Christian Marshall for annotation of variants with Human Gene Mutation Database; and Susan Walker and Stephen W. Scherer for providing variant calls in unaffected parents from an Ontario cohort with autism spectrum disorders. The work was supported by funding from the Heart and Stroke Foundation of Ontario Chair in Cardiovascular Science (S.M.), Heart and Stroke Foundation of Canada Research Fellowship (A.K.M.), McLaughlin Centre Accelerator Grant in Genomic Medicine (S.M.), SickKids Labatt Family Heart Centre Innovation Funds (S.M., L.C.A.D., A.K.M., and D.M.), the Roma and Marvin Auerback Endowment funds (S.M.), Wellcome Trust (grant WT098051; M.E.H., S.A.T.), British Heart Foundation (grants CH/09/003/26631 and RG/10/17/28553; S.B. (Shoumo Bhattacharya)).
Members of the UK10K Rare Diseases Cohorts Working Group are Matthew Hurles (co-chair), David R. FitzPatrick (co-chair), Saeed Al-Turki, Carl Anderson, Ines Barroso, Philip Beales, Jamie Bentham, Shoumo Bhattacharya, Keren Carss, Krishna Chatterjee, Sebhattin Cirak, Catherine Cosgrove, Allan Daly, Jamie Floyd, Chris Franklin, Marta Futema, Steve Humphries, Shane McCarthy, Hannah Mitchison, Francesco Muntoni, Alexandros Onoufriadis, Victoria Parker, Felicity Payne, Vincent Plagnol, Lucy Raymond, David Savage, Peter Scambler, Miriam Schmidts, Robert Semple, Eva Serra, Jim Stalker, Margriet van Kogelenberg, Parthiban Vijayarangakannan, Klaudia Walter, and Gretta Wood.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Supplementary information
Supplementary Figure S1
(TIFF 2778 kb)
Supplementary Figure S2
(TIFF 166 kb)
Supplementary Figure S3
(TIFF 223 kb)
Supplementary Figure S4
(TIFF 1722 kb)
Supplementary Figure S5
(TIFF 3328 kb)
Supplementary Tables
(DOC 459 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
D’Alessandro, L., Al Turki, S., Manickaraj, A. et al. Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet Med 18, 189–198 (2016). https://doi.org/10.1038/gim.2015.60
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.60
Keywords
This article is cited by
-
Predisposition to atrioventricular septal defects may be caused by SOX7 variants that impair interaction with GATA4
Molecular Genetics and Genomics (2022)
-
Congenital heart defects among Down’s syndrome cases: an updated review from basic research to an emerging diagnostics technology and genetic counselling
Journal of Genetics (2021)
-
Genetics of atrioventricular canal defects
Italian Journal of Pediatrics (2020)
-
A familial congenital heart disease with a possible multigenic origin involving a mutation in BMPR1A
Scientific Reports (2019)
-
Targeted Next-Generation Sequencing in Patients with Non-syndromic Congenital Heart Disease
Pediatric Cardiology (2018)
