
Abstract
Purpose:
Chromosomal microarray analysis is the gold standard for copy-number variant (CNV) detection in prenatal and postnatal diagnosis. We aimed to determine whether next-generation sequencing (NGS) technology could be an alternative method for CNV detection in routine clinical application.
Methods:
Genome-wide CNV analysis (>50 kb) was performed on a multicenter group of 570 patients using a low-coverage whole-genome sequencing pipeline. These samples were referred for chromosomal analysis; CNVs (i.e., pathogenic CNVs, pCNVs) were classified according to the American College of Medical Genetics and Genomics guidelines.
Results:
Overall, a total of 198 abortuses, 37 stillbirths, 149 prenatal, and 186 postnatal samples were tested. Our approach yielded results in 549 samples (96.3%). In addition to 119 subjects with aneuploidies, 103 pCNVs (74 losses and 29 gains) were identified in 82 samples, giving diagnostic yields of 53.2% (95% confidence interval: 45.8, 60.5), 14.7% (5.0, 31.1), 28.5% (21.1, 36.6), and 30.1% (23.6, 37.3) in each group, respectively. Mosaicism was observed at a level as low as 25%.
Conclusions:
Patients with chromosomal diseases or microdeletion/microduplication syndromes were diagnosed using a high-resolution genome-wide method. Our study revealed the potential of NGS to facilitate genetic diagnoses that were not evident in the prenatal and postnatal groups.
Genet Med 18 9, 940–948.
Similar content being viewed by others

Introduction
DNA copy-number variants (CNVs) account for up to 300 Mb of sequence variation in a normal human individual, with a variety of lengths of up to millions of base pairs.1,2 This represents the major genome diversity for two different individuals; some of these CNVs are known to be associated with the pathogenicity of a variety of human disorders, including the commonly known DiGeorge syndrome (OMIM 88400),3 Angelman syndrome (OMIM 105830),4 and neurogenetic defects such as ATR syndrome (16p deletion; OMIM 610543, 613604, and 613444).5
To detect pathogenic CNVs, chromosomal microarray analysis (CMA), including array–comparative genomic hybridization (array-CGH)6,7 and single-nucleotide polymorphism array,8,9 has been widely used as a gold standard. Compared with CMA, next-generation sequencing (NGS) is an alternative state-of-the-art technology promising improved detection of genetic abnormalities with unprecedented resolution.10 Recently, a few retrospective studies with limited sample size have supported the performance of NGS for detecting CNVs in clinical samples.11,12,13,14
To study the diagnostic effectiveness and feasibility of using a low-coverage (or low-pass) whole-genome NGS approach to detect chromosomal numerical and structural abnormalities in a diagnostic laboratory, we applied our in-house CNV detection method for a multicenter group of 570 patients referred to chromosomal analysis. A total of 198 abortuses, 37 stillbirths, 149 prenatal samples, and 186 postnatal samples were tested.
Materials and Methods
Subject enrollment and sample recruitment
The study was approved by the institutional review board of each collaborative site. Written consent for storage and subsequent analysis was obtained from each participant. Products of conception from first-trimester miscarriage and fetal tissue from stillbirths were collected. For prenatal samples, chorionic villi, amniotic fluid, and cord blood were collected. A peripheral blood sample was collected from each postnatal patient who was phenotypically abnormal and referred for genetic testing. The YH lymphocytic cell line (the first Asian human genome sequenced)15 was also used for methodology evaluation.
Sample preparation for NGS
DNA was extracted with the use of a commercial DNA-extraction kit (Puregene; Qiagen, Hilden, Germany) and then quantified with the Qubit dsDNA HS Assay Kit (Invitrogen, Life Technologies, Waltham, MA) for DNA quality control (QC) measurement. All samples passing QC (>500 ng; OD260/OD280 > 1.8; OD260/OD230 > 1.5) were subsequently prepared for a non-size-selected library (~250 bp) protocol; in brief, 100 ng of genomic DNA was sheared into small fragments (200–300 bp) with Covaris S2 (Covaris, Woburn, MA). After end repair, addition of an A overhang and adapter ligation, DNA fragments (without size selection) underwent 10 cycles of polymerase chain reaction (PCR). For the samples with size-selection library construction, 500 ng of genomic DNA from each sample was first sheared into small (400–600 bp) fragments with Covaris S2. After end repair, addition of an A overhang and adapter ligation, 12 cycles of PCR were carried out using the DNA fragments with adapter molecules at both ends. The size-selection (550–650 bp) procedure for the PCR products was performed via 2% agarose gel electrophoresis. Gel slices were excised and purified with a QIAquick Gel Extraction kit (Qiagen) following the manufacturer’s protocol.
PCR products from each library were subsequently purified with an Agencourt AMPure XP PCR Purification Kit (Beckman Coulter, Brea, CA). The size distribution of each library was assessed using a 2100 Bioanalyzer DNA 1000 kit (Agilent Technologies, Santa Clara, CA). Concentrations of the PCR products were measured by quantitative PCR (qPCR). Non-size-selected libraries with different index tags with equal molality were mixed into a pool (12 or 24 samples per lane) and sequenced with 50-base single-end sequencing (~15 million reads per sample) on a HiSeq 2000 platform (Illumina, San Diego, CA). Each size-selected library was 50-base paired–end sequenced with ~90 million read pairs (2 samples per lane), also on the HiSeq 2000 platform (Illumina).
Read-depth estimation, which is used to determine a sufficient depth of coverage for a target region before further analysis, was performed for each sample. It is calculated by multiplying the read amount produced (~15 million) and the read length (50-base single-end sequencing) and dividing the result by the target size (3 Gb as the size of the whole human genome). Therefore, for each sample with a non-size-selected library for CNV analysis, a read depth of ~0.25× was produced in this study.
CNV analysis
Reads were aligned to the National Center for Biotechnology Information human reference genome (hg19, GRCh37.1, hereafter called hg19) using SOAP2 (ref. 16), and PCR duplicates were removed. Only uniquely mapped reads were selected.13 Apart from aneuploidy diagnosis17 (Supplementary Methods online), in general, CNV detection was performed according to the three steps below:
-
1. Quality control and putative CNVs screened with sliding windows. Mapped reads were classified into adjustable sliding windows,18,19 which were 50 kb in length with 5 kb increments (Supplementary Methods and Supplementary Figure S1a online), in terms of their mapped locations (hg19). The coverage of each window was calculated by the read amount and underwent two-step bias correction13 (GC correction and population-scale normalization) (Supplementary Methods online). For the QC step, we first excluded the windows located in the chromosomes of numerical disorders and then calculated the genome-wide standard deviation (GWSD) of the windows’ copy ratios. After two-step correction, the sample is considered to have passed QC if the GWSD is <0.15, which is twice the GWSD of YH (Supplementary Figure S2 online). For samples that passed QC, candidate CNV regions genome-wide were screened and flagged for subsequent analysis (Supplementary Methods online).
-
2. Precise boundaries identified with increment rate of coverage of the adjustable nonoverlapping windows. For more precise identification of CNV boundaries (windows), the aligned reads were also classified into nonoverlapping windows. After that, for any particular adjustable nonoverlapping window18 (5 kb), the increment ratio of coverage (Figure 2a and Supplementary Figure S3 online) was calculated as the coverage difference divided by its coverage. To detect the most precise boundaries of altered copy-number regions, we used circular peak–trough screening (Supplementary Methods and Supplementary Figure S3 online) and considered the average copy number of the newly determined “segment” or region.
-
3. Individual CNV annotation and interpretation. A CNV is defined as a deletion when its average copy ratio is less than 0.9 (mosaics: 0.6 to 0.9) or as a duplication when greater than 1.1 (mosaics: 1.1 to 1.4), if this event is an outlier as determined by a U-test (P value <0.0001) from a normal population with 100 samples13 (the 1000 Genomes Project). Classification of CNVs is based on the American College of Medical Genetics and Genomics guidelines,20 with the criteria summarized below:
-
a. Pathogenic or likely pathogenic CNV: (i) contains a pathogenic autosomal dominant gene defined by GeneReviews; (ii) harbors 50% of the length of a critical region of a known syndrome defined in DECIPHER; (iii) covers the full length of pathogenic CNVs defined by ClinVarCNV or (iv) contains gene(s) reported in both OMIM and HGMD.
-
b. Variant of uncertain significance (VOUS, hereafter referred to as CNV without further subclassification) is classified as (i) covering the full length of a VOUS defined by ClinVarCNV, or (ii) deleted and contains gene(s) reported either in OMIM or HGMD only, or (iii) contains genes, but it is not known whether the genes in the interval are dosage-sensitive by CMA-based databases (ClinVarCNV, DECIPHER, and in-house databases from Baylor College of Medicine and The Chinese University of Hong Kong).
-
CMA
We used a well-established customized 44K Fetal DNA Chip v1.0 (Agilent Technologies)7 and Human CytoSNP-12 BeadChip with 300,000 probes (Illumina)21 for CMA. Single-nucleotide polymorphism array and array-CGH tests were performed according to the manufacturers’ protocols. For array-CGH, CNVs were analyzed via CytoGenomics.7 For single-nucleotide polymorphism array, CNV analysis was carried out using KaryoStudio V 1.3.11 and GenomeStudio V2011.1 (ref. 21) in parallel.
Multiplex ligation-dependent probe amplification validation
Probes (Supplementary Table S4 online) were designed from the unique sequences within the CNV regions using RAW software ((MRC-Holland BV, Amesterdam, the Netherlands).22 Synthetic probes were diluted to a final concentration of 4 nmol/l, and 0.5 µl was added to the P300 (MRC-Holland BV) probe mix. After hybridization, ligation, amplification, and electrophoresis with a standard multiplex ligation-dependent probe amplification procedure, data were collected with a 3100 sequencer (ABI, Life Technologies) and analyzed with Gene Marker 1.9 software (Softgenetics, State College, PA).
Accession number
Whole-genome sequencing reads are available in the National Center for Biotechnology Information Sequence Read Archive under accession number SRA296708.
Code availability
All the programs relevant to this pipeline are available at http://sourceforge.net/projects/increment-ratio-of-coverage/files/Increment_Ratio_of_Coverage.tar.gz/download.
Results
To evaluate the performance of our in-house whole-genome low-coverage sequencing–based approach, we used DNA samples with known CMA results for comparison. Sixty-eight DNA samples (51 prenatal and 17 postnatal samples) with various CNVs as well as 3 prenatal samples with mosaic pathogenic CNVs (pCNVs) were selected for NGS testing (~15 million reads or ~0.25× per sample) in blinded fashion, and the detection results were further compared with the CMA reports ( Table 1 ).
In addition to identifying numerical abnormalities (Supplementary Table S1 online), our approach identified 42 constitutional pCNVs ranging from 1.3 to 69.1 Mb in the prenatal group ( Table 1 , Supplementary Table S1 online, and Figure 1a ), results that were 100% consistent with the CMA reports. In the postnatal group, our method identified nine constitutional pCNVs ranging from 1.6 to 20.6 Mb, which were also confirmed by CMA ( Table 1 , Supplementary Table S1 online, and Figure 1a ). For constitutional pCNV detection, the NGS-based method provided 100.0% (91.0, 100.0) sensitivity and 100.0% (89.1, 100.0) specificity.

CNV detection in a validation group and a multicenter clinical group. (a) The distribution of CNVs (including trisomy 18 detected in sample 12DNJ067) by CMA (inner circle) and our NGS-based approach (outer circle) in 71 validation samples. Karyotypical structures and cytogenetic band colors are shown according to the University of California, Santa Cruz Genome Viewer Table Browser and chromosome color schemes (outmost circle). Rectangles in red and blue indicate consistent copy-number losses and gains, respectively, detected by both methods. Furthermore, for the CNVs uniquely called by CMA (inner circle) or NGS (outer circle), losses and gains are shown in purple and in green, respectively. Three deletions (4q34.3q35.2, 5p15.33 and 15q11.2), two duplications (1q25.2q44 and 17p13.3) and a triplication (12p11.1p13.33) in mosaic fashion are shown (arrows) with mosaic level (percentage). (b) The distribution of pathogenic or likely pathogenic CNVs detected in a multicenter group including 198 early abortuses, 37 stillbirths, 149 prenatal samples, and 186 postnatal samples. Rectangles in red and blue indicate the pathogenic copy-number losses and gains, respectively. CMA, chromosomal microarray analysis; CNV, copy-number variant; NGS, next-generation sequencing.
For mosaic CNV detection, our NGS-based approach detected six mosaics, four of which were pCNVs ( Figure 1a and Supplementary Table S1 online), consistent with the three samples indicated with mosaic pCNVs by CMA. In sample HK12C0637, there was a mosaic copy-number gain of 2.9 Mb at about the 40% level located at 17p13.3(698057-3593612) × 2–3 ( Figure 2a , b and Supplementary Table S1 online) next to a combination of a constitutional deletion (167.0 kb) and a duplication (517.5 kb). In sample HK12C0310, our approach identified a mosaic deletion of 171.0 kb at 5p15.33(684118–855103) × 1–2 and a mosaic triplication of 34.8 Mb at 12p11.1p13.33(60105-34836577) × 3–4, both at about the 50% level (Supplementary Table S1 online). For the third sample, named HK12C0669, a terminal mosaic duplication was identified at 1q25.2q44(179647508-249171049) × 2–3, while a terminal mosaic deletion was identified at 4q34.3q35.2(177543828-190910498) × 1–2, both at about the 50% level (Supplementary Table S1 online). Overall, 100% concordance between our NGS pipeline and CMA was achieved for chromosomal numeric and pathogenic CNV detection, indicating that the sensitivity and specificity of pCNV (constitutional and mosaic) detection were 100.0% (91.4, 100.0) and 100.0% (88.4, 100.0), respectively, for our NGS-based approach.

Validation of CNV detection by MLPA and CMA. (a) Constitutional deletion and duplication, followed by mosaic copy-number gain detected by NGS in sample HK12C0637, which were confirmed by (b) CMA. C.Del., constitutional deletion; C.Dup., constitutional duplication; Mos.Dup., mosaic duplication. Numbers in parentheses in a indicate the copy ratio of the DNA fragment. Numbers in b show the log2ratio provided by Agilent’s Cytogenomics software. Percentage reflects the mosaic level detected by our NGS-based approach. (c) Identification of the precise region of duplication located in 7q36.2 (153476694-153660423) × 3 in sample 12ZS121913. (d) MLPA validation reflects the increased dosage of 7q36.2. (e) NGS and (f) CMA results show consistent mosaic trisomy 2 (percentage: 40–50%) in sample 14S1003400. In a and c, black lines indicate the distribution of copy ratios for adjusted nonoverlapping windows (5 kb), and orange lines indicate the distribution of increment rate of coverage. In b and f, dots in red, blue, and black indicate the copy-ratio loss, gain, and eusomic, respectively. CMA, chromosomal microarray analysis; CNV, copy-number variant; MLPA, multiplex ligation-dependent probe amplification; NGS, next-generation sequencing.
We further selected 14 samples described above with sufficient DNA for low-coverage paired-end sequencing to fine-map the copy-number regions identified by our established method23 (Supplementary Figure S4a online). Using simulation in the YH lymphocytic cell line15 (Supplementary Table S2 online) or anomalous read pairs detected by our balanced chromosomal rearrangement (or balanced chromosomal abnormality) pipeline23 (Supplementary Table S3 online), we were able to identify both the start and stop locations of each CNV within an adjusted nonoverlapping window of ~5 kb from the precise region compared with an interval of ~21.5 kb by CMA (inner boundaries, Supplementary Table S3 online). Our approach detected 32 additional CNV events as VOUS compared with CMA ( Table 1 ), and such additional information may in turn provide more important information for disease gene(s) discovery. For instance, patient 12ZS121913 with DiGeorge syndrome (OMIM 188400)3 diagnosed by clinical phenotype was defined as chromosomally normal by CMA, but our approach detected a copy-number gain of 183.7 kb located at 7q36.2(153476694-153660423) × 3 ( Figure 2c ) containing DPP6, overexpression of which has been demonstrated to be associated with heart disease (i.e., familial idiopathic ventricular fibrillation).24 Multiplex ligation-dependent probe amplification validation22 confirmed this copy-number gain ( Figure 2d and Supplementary Table S4 online), which was missed by CMA due to the probe design (Supplementary Figures S5 and S6 online). The same scenario was found in sample 12DNJ063 with a congenital heart disorder; in this patient, a 107.5 kb deletion was detected at 20p12.1(14818084-14925606) harboring gene MACROD2. It is within an intron or involves some noncoding regions in some transcripts of MACROD2, and heart disease is not a consistent feature in DECIPHER cases with overlapping deletions in the region. However, mutation in this gene has been reported to be associated with heart disease.25
After evaluating the sensitivity and specificity of the NGS-based molecular karyotyping in pCNV detection, we implemented this platform in our routine diagnostic laboratory setting to further evaluate its performance. We then obtained 570 samples from four tertiary referral centers in China and Hong Kong from January 2013 to March 2015. These samples included 198 abortuses, 37 stillbirths, 149 prenatal samples, and 186 postnatal samples.
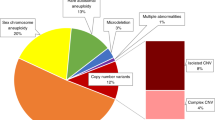
Sequence-based analysis was successful in 549 samples (96.3%, Figure 1b ). Among failed cases (21/570), 10 were abortuses from early pregnancies, 3 were stillbirths, and the other 8 were induced terminations with ultrasound anomalies. They all had poor DNA quality, most likely due to fetal demise. Overall, we identified a total of 2,411 CNVs (790 losses and 1,621 gains) in the 549 samples studied. Aneuploidies were identified in 119 samples, and 104 pCNVs (74 losses and 29 gains) were detected in 82 samples, giving an overall diagnostic yield of 36.6% (32.6, 40.8; Table 2 ). In this group, NGS-based molecular karyotyping identified 11 cases of mosaic aneuploidies, of which the majority were early abortuses (10/11, Supplementary Table S5 online); maternal cell contamination was excluded in all cases by quantitative fluorescence–polymerase chain reaction.26
Of the 549 successful cases, we “randomly” selected 25 from 368 samples with sufficient DNA quantity (minimum 500 ng) for validation using CMA. These samples included 5 aneuploidies (3 mosaics, Figure 2e , f and Supplementary Table S5 and Supplementary Figure S7a,b online) and 13 pCNVs (Supplementary Tables S6–S10 and Supplementary Figure S7c,d online) within regions with probe coverage. All these events were 100% consistent with CMA, indicating a high level of consistency and robust performance of the NGS platform.
Diagnostic yield among spontaneous abortions and stillbirths
Chorionic villi or placental tissues from 198 abortuses, which were miscarried in the first trimester of pregnancy, were collected for DNA extraction and subsequent sequencing. Ten samples failed QC (5.1%, Supplementary Methods online). Of the 188 samples, 72 had a single aneuploidy (38.3%); among them, 19 samples had sex-chromosome aneuploidy, Supplementary Table S5 online). In addition, six samples (3.2%) were diagnosed with more than one chromosome aneuploidy (Supplementary Table S5 online). Twelve samples (6.4%) were determined to have 15 pCNVs (Supplementary Table S6 online), ranging from 349.4 kb to 78.2 Mb, and 13 pCNVs were larger than 1 Mb.
A total of 10 samples were interpreted as autosomal aneuploidy in mosaic fashion (Supplementary Table S5 online and Figure 2c ). The overall diagnostic yield in the early abortus group was 53.2% (100/188, 45.8, 60.5) and the incidence of chromosomal numerical abnormalities was 46.8% (39.5, 54.2).
Fetal tissues (N = 35) and amniotic fluids (N = 2) from 37 stillbirths were studied. Three samples failed the QC check (8.1%). Of the 34 samples, three (8.8%) with aneuploidy (two samples with sex-chromosome aneuploidy) were detected (Supplementary Table S5 online). Two samples (5.9%) were found to have three pCNVs (Supplementary Table S7 online). The overall detection rate in this sample type was 14.7% (5/34, 5.0, 31.1).
Diagnostic yield in the prenatal group
Thirty-six amniotic fluid samples, 18 cord blood samples referred from high-risk pregnancies, and 95 aborted fetal samples (muscle, chorionic villus, or placental tissue) with ultrasound anomalies were recruited. Eight samples failed QC testing (5.4%). Of the 141 samples, 20 (13.5%) had numerical abnormalities, of which 6 were sex-chromosome aneuploidies (Supplementary Table S5 online). Nineteen samples (13.5%) were diagnosed as having 23 pCNVs (Supplementary Table S8 online) ranging from 59.6 kb to 31.3 Mb, of which 17 were larger than 1 Mb. For samples with parallel conventional karyotyping, all results were consistent with our NGS detection (Supplementary Table S8 online). In addition, sample 15B6111364 was identified as a mosaic for monosomy 13 (Supplementary Table S5 online), but no culture sample was available for confirmation by fluorescence in situ hybridization confirmation. In total, the detection rate of chromosomal abnormality in the prenatal sample type was 28.4% (40/141, 21.1, 36.6), and, excluding aneuploidies, the yield was 13.5% (8.3, 20.3).
Diagnostic yield in the postnatal group
One hundred eighty-six whole-blood DNA samples were collected from individuals ranging from 1-day-old newborns to adults with phenotypic anomalies, including 12 couples (24 samples) requesting genetics diagnosis and counseling with a history of pregnancy losses of fetuses with ultrasound anomalies. Of the 186 samples, 7 were identified as common trisomy syndromes (3.8%, Supplementary Table S5 online). In 49 samples (26.3%, Supplementary Table S9 online), 62 pCNVs were detected, ranging from 60.9 kb to 41.7 Mb. Three pairs of parental DNAs were recruited for three probands with pCNVs, and two were diagnosed as de novo (Supplementary Table S9 online). The third sample, 13U0252513, was suspected to be an unbalanced segregant of a parental balanced translocation because it had a deletion and a duplication in the terminal regions of two chromosomes. To confirm this event, we applied our balanced chromosomal abnormality detection method to each member from this trio and identified chimeric read pairs supporting the proband’s karyotype as der(5)t(5;12)(p14.2;p13.1)pat ( Figure 3a ). This translocation was also confirmed by conventional karyotyping ( Figure 3b , c ). In addition, a family member of sample 14B0037388 with the same phenotypic anomalies was referred for testing. The proband, a 6-year-old girl (14B0037388), and her 3-year-old younger brother (14B0593365) who presented with intellectual disability and speech delay, were found to have a common deletion in 22q13 (ref. 27), most likely due to an unbalanced segregant of a parental balanced translocation. Interestingly, sample 14B0593391, a phenotypically normal male subject, displayed a deletion of 113.0 kb (classified as a VOUS) in 6q26(162800625-162913644)x1 that contained PARK2, which is related to neural development.28 His wife had three pregnancies with hydrocephalus; however, no DNA samples were obtained to confirm inheritance to the hydrocephalic fetuses.

Composition of the derivative chromosomes in a balanced translocation detected by NGS and confirmed by conventional karyotyping. (a) Joining sequences of two derivative chromosomes in a paternal carrier. Precise breakpoints (shown in dotted line) are located in 12p13.1 (14,780,019, based on hg19) and 5p14.2 (23,314,435) with two bases of microdeletion (characters in red) and microhomology (characters in blue), respectively, which were confirmed by Sanger sequencing; (b and c) conventional G-banded partial karyotypes showing t(5;12)(p14.2;p13.1) in father and der(5)t(5;12)(p14.2;p13.1)pat in the proband. CNV, copy-number variant; NGS, next-generation sequencing.
In total, the detection rate of chromosomal abnormalities in this postnatal sample type was 30.1% (56/186, 23.6, 37.2), providing an additional diagnostic yield of 12.8% (19/149, 7.9, 19.3) among karyotypically normal subjects. This is based on the assumption that pathogenic CNV size less than 5 Mb could not be detected by conventional cytogenetic analysis.
Discussion
This study was designed to determine whether it would be more efficient to perform an NGS-based approach rather than conventional CMA. In our validation group, our NGS approach was 100% consistent with CMA analysis for constitutional and mosaic pCNVs detection. Furthermore, 32 additional VOUS were detected because of a more evenly distributed approach (whole-genome sequencing analysis) and a finer resolution of our algorithm (50 kb) compared with probe selection by CMA.
In our clinical group, our NGS approach reached a diagnostic yield of 36.6% (201/ 549, 32.6, 40.8), indicating 1 in 2.7 chromosomal numerical and/or structural anomalies among those referred for genetic diagnosis. This high detection rate was due primarily to the much higher diagnostic yield derived from the group of spontaneous abortion than that from other sample types. However, this group in the current study is a representative group in clinical diagnostic labs where chromosomal microdeletion/microduplication analysis has been well applied. For the stillbirth samples, owing to the small sample size in this group (N = 34), sample bias may have contributed to the higher diagnostic yield that was observed, compared with recently published studies.29,30 Nonetheless, our current data truly show a higher diagnostic yield. For the prenatal and postnatal samples, our NGS approach gave a much better diagnostic yield than conventional karyotyping analysis in a tertiary referral center.7,31,32 Apart from aneuploidy detection, the prevalence of pathogenic or likely pathogenic CNVs was 6.4% (3.3, 10.9), 5.9% (0.7, 19.7), 13.5% (8.3, 20.3), and 26.3% (20.1, 33.2) in early abortuses, stillbirths, prenatal samples, and postnatal samples, respectively. These are comparable to the diagnostic yields detected by CMA,7,29,30,31,33,34,35 indicating that our detection rate was consistent with the sample group/type.
The median size of pCNVs (N = 104) was ~5.0 Mb, demonstrating the clinical utility of NGS in molecular diagnostics. In addition, based on CNV classification as recommended by the American College of Medical Genetics and Genomics,20 467 CNVs (164 losses and 303 gains) were defined as VOUS ranging from 50.2 kb to 2.5 Mb (Supplementary Figure S8 online). Our NGS-based approach identified 1,823 benign or likely benign CNVs in our clinical groups (549 losses and 1,274 gains) as defined by CMA-based databases. On average, each sample was reported to carry 3.3 benign variants (1.0 loss and 2.3 gains). Among them, 98.2% were smaller than 1 Mb, compared with 93.5% in the ClinVarCNV database of benign CNVs. Overall, there was a significant difference in CNV size between VOUS and benign CNVs in this group (two-sample Kolmogorov-Smirnov test: P = 0.0041, considered significant at P < 0.05, Supplementary Figure S8 online). Therefore, a comprehensive database including data generated from sequencing-based methods for studying the pathogenicity of VOUS will be useful. Our method employed a population-based U-test for filtering out CNVs with high frequency in the control group or directly normalized by the control baseline. The reason that there are still benign CNVs reported as VOUS is their low incidence (<5%) in normal populations, requiring further classification with CMA-based or even NGS-based databases.
It is difficult to identify the phenotype of a pCNV detected in an early abortus, stillbirth, or prenatal sample. In this scenario, we can use the postnatal-based database for interpretation of the clinical significance or prognosis. For instance, prenatal sample 14S0026471 was referred for testing because of an ultrasound anomaly identified at 23 weeks of gestation. A deletion of 1.3 Mb located at 15q13.2q13.3(31113471-32418619)x1 was detected and classified as likely pathogenic because this deletion has been implicated in the 15q13.3 deletion syndrome (OMIM 612001), which has been reported in postnatal cases with typical features (e.g., intellectual disability and seizures).36
Because most well-known syndromes have been characterized as harboring one or more disease-causing genes, identification of the precise boundaries of copy-number changes as detected by our NGS platform is important for defining a new syndrome. In this study, our approach is demonstrated to be more precise in identifying the critical region by our newly established increment ratio of coverage algorithm. Compared to CMA, which is limited by probe spacing and density, our NGS method is more accurate and precise for mapping the critical region of diseases.
For chromosomal rearrangement, nine samples (9/549) were noted to have deletion and duplication in the terminal q/p arm of one or more chromosomes simultaneously, implicating unbalanced translocations (Supplementary Table S10 online). In NGS read-depth–based methods,37 only dosage changes, rather than the real composition of derivative chromosome(s), can be observed. Therefore, karyotyping is recommended for investigation of unbalanced translocations because of the spatial visualization. However, three samples had a terminal deletion or duplication at a submicroscopic level (<5 Mb), unlikely to be detectable by conventional karyotyping but detectable by our established balanced chromosomal abnormality detection method.23
For prenatal samples obtained from invasive testing, the turnaround time is 10 days from sample receipt to diagnostic report, which is competitive with CMA. Based on our data using the HiSeq 2000 platform, it is possible to evaluate sequence information for up to 96 samples in a single sequencing slide (two per run), which may ultimately lead to reducing the costs per patient. Based on the reagent costs including (i) DNA extraction (including DNA QC measurement), (ii) library construction (non-size-selected), and (iii) about 15 million single-end reads in the HiSeq 2000 platform, costs are estimated at about US$120 per sample. Given a staff cost as $50 per hour, as each lane/run requires 16 working hours, the labor cost is estimated to be $67 per sample (e.g., 12 samples per lane). Therefore, in total, the cost for each sample would be about $187, which compares favorably to the cost of conventional karyotyping. In addition, it allows a more extensive numerical and pCNV detection in various sample types, particularly in prenatal diagnosis, compared with the current standard of CMA.
A limitation of this NGS-based CNV detection is, as with CMA, a requirement for high-quality DNA for testing. Therefore, this method may not be fully applicable for DNA samples extracted from a fetal demise29 (6.4%, 21/328 failed in our study). Moreover, neither array-CGH nor low-coverage whole-genome sequencing with a read-depth CNV-detection algorithm can detect triploidy.38 In this study, an early abortus sample (ID 14S0026197) was identified by single-nucleotide polymorphism array as 69,XXX but was not detected by our assay, which represents a limitation of our method. Furthermore, triploid fetuses often result in early spontaneous abortions and would hence go undetected.38 With the exception of the above limitations, our approach provides a high-throughput, robust, genome-wide high-resolution analysis pipeline for detection of numerical disorders and CNVs (particularly for pCNV).
Compared with CMA, in the clinical diagnosis of aneuploidy and pCNVs, our NGS-based approach shows equivalent effectiveness and advantages, including the detection of chromosomal mosaicism at a low level. In this study, 11 samples displayed mosaicism that ranged from 25 to 70%. The NGS-based approach has been reported to detect lower-percentage mosaics in other sample types (maternal plasma),39 indicating the potential to detect accurately lower levels of mosaicism chromosomal abnormalities in our sample types. Second, our NGS approach provides additional genome-wide detection of pCNVs or VOUS compared with CMA. In the validation group, our approach revealed 32 additional CNVs as VOUS, which may provide important information for gene discovery ( Figure 2 ). Finally, the high success rate in our NGS-based approach demonstrates a high diagnostic rate in fetal demise40 (93.6%), compared with 87.4% in a microarray study on stillbirth.29
In summary, chromosomal diseases or microdeletion/microduplication syndromes can be diagnosed effectively by NGS. Our study demonstrated that NGS is a robust, sensitive, and high-resolution genome-wide method to identify numerical and pathogenic CNVs among prenatal and postnatal patients. Furthermore, our study highlights the potential for using NGS to facilitate genetic diagnoses in the prenatal and postnatal samples that have not been detected by conventional karyotyping and/or CMA analysis.
Disclosure
The authors declare no conflict of interest.
References
Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65.
Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009;10:241–251.
Jonas RK, Montojo CA, Bearden CE. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry 2014;75:351–360.
Greer PL, Hanayama R, Bloodgood BL, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010;140:704–716.
Lamb J, Harris PC, Wilkie AO, Wood WG, Dauwerse JG, Higgs DR. De novo truncation of chromosome 16p and healing with (TTAGGG)n in the alpha-thalassemia/mental retardation syndrome (ATR-16). Am J Hum Genet 1993;52:668–676.
Tang YC, Amon A. Gene copy-number alterations: a cost-benefit analysis. Cell 2013;152:394–405.
Leung TY, Vogel I, Lau TK, et al. Identification of submicroscopic chromosomal aberrations in fetuses with increased nuchal translucency and apparently normal karyotype. Ultrasound Obstet Gynecol 2011;38:314–319.
Pinto D, Darvishi K, Shi X, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 2011;29:512–520.
Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet 2005;37:549–554.
Mills RE, Walter K, Stewart C, et al.; 1000 Genomes Project. Mapping copy number variation by population-scale genome sequencing. Nature 2011;470:59–65.
Lui S, Song L, Cram DS, et al. Traditional karyotyping vs copy number variation sequencing for detection of chromosomal abnormalities associated with spontaneous miscarriage. Ultrasound Obstet Gynecol 2015;46:472–477.
Liang D, Peng Y, Lv W, et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J Mol Diagn 2014;16:519–526.
Li X, Chen S, Xie W, et al. PSCC: sensitive and reliable population-scale copy number variation detection method based on low coverage sequencing. PLoS One 2014;9:e85096.
Duan J, Zhang JG, Deng HW, Wang YP. Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS One 2013;8:e59128.
Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature 2008;456:60–65.
Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009;25:1966–1967.
Xie W, Tan Y, Li X, et al. Rapid detection of aneuploidies on a benchtop sequencing platform. Prenat Diagn 2013;33:232–237.
Szatkiewicz JP, Wang W, Sullivan PF, Wang W, Sun W. Improving detection of copy-number variation by simultaneous bias correction and read-depth segmentation. Nucleic Acids Res 2013;41:1519–1532.
Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008;18:1851–1858.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST ; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–685.
Hu P, Meng L, Ma D, et al. A novel 11p13 microdeletion encompassing PAX6 in a Chinese Han family with aniridia, ptosis and mental retardation. Mol Cytogenet 2015;8:3.
Eijk-Van Os PG, Schouten JP. Multiplex ligation-dependent probe amplification (MLPA®) for the detection of copy number variation in genomic sequences. Methods Mol Biol 2011;688:97–126.
Dong Z, Jiang L, Yang C, et al. A robust approach for blind detection of balanced chromosomal rearrangements with whole-genome low-coverage sequencing. Hum Mutat 2014;35:625–636.
Alders M, Koopmann TT, Christiaans I, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet 2009;84:468–476.
Slavin TP, Feng T, Schnell A, Zhu X, Elston RC. Two-marker association tests yield new disease associations for coronary artery disease and hypertension. Hum Genet 2011;130:725–733.
Cheng YK, Wong C, Wong HK, et al. The detection of mosaicism by prenatal BoBs™. Prenat Diagn 2013;33:42–49.
Wilson HL, Wong AC, Shaw SR, et al. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet 2003;40:575–584.
Liang F, Li W, Zhang P, et al. A PARK2 polymorphism associated with delayed neuropsychological sequelae after carbon monoxide poisoning. BMC Med Genet 2013;14:99.
Reddy UM, Page GP, Saade GR, et al.; NICHD Stillbirth Collaborative Research Network. Karyotype versus microarray testing for genetic abnormalities after stillbirth. N Engl J Med 2012;367:2185–2193.
Sahlin E, Gustavsson P, Liedén A, et al. Molecular and cytogenetic analysis in stillbirth: results from 481 consecutive cases. Fetal Diagn Ther 2014;36:326–332.
Chong WW, Lo IF, Lam ST, et al. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet 2014;7:34.
Bruno DL, Ganesamoorthy D, Schoumans J, et al. Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J Med Genet 2009;46:123–131.
Liu J, Bernier F, Lauzon J, Lowry RB, Chernos J. Application of microarray-based comparative genomic hybridization in prenatal and postnatal settings: three case reports. Genet Res Int 2011;2011:976398.
Lathi RB, Massie JA, Loring M, et al. Informatics enhanced SNP microarray analysis of 30 miscarriage samples compared to routine cytogenetics. PLoS One 2012;7:e31282.
Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–2184.
Sharp AJ, Mefford HC, Li K, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet 2008;40:322–328.
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics 2013;14(suppl 11):S1.
McFadden DE, Robinson WP. Phenotype of triploid embryos. J Med Genet 2006;43:609–612.
Wallerstein R, Misra S, Dugar RB, Alem M, Mazzoni R, Garabedian MJ. Current knowledge of prenatal diagnosis of mosaic autosomal trisomy in amniocytes: karyotype/phenotype correlations. Prenat Diagn 2015;35:841–847.
Grati FR, Gomes DM, Ganesamoorthy D, et al. Application of a new molecular technique for the genetic evaluation of products of conception. Prenat Diagn 2013;33:32–41.
Acknowledgements
This study was approved by the National Basic Research Program of China (2012CB944600), the Shenzhen Municipal Commission for Development and Reform and Key Laboratory Project in Shenzhen (CXB200903110066A and CXB201108250096A), the National Natural Science Foundation of China (81300495), the Medical Leading Talent and Innovation Team Project of Jiangsu Province (LJ201109), and the Key Technology R&D Program of Jiangsu Province (BL2012039 and F201314).
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
(PDF 3516 kb)
Rights and permissions
About this article

Cite this article
Dong, Z., Zhang, J., Hu, P. et al. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med 18, 940–948 (2016). https://doi.org/10.1038/gim.2015.199
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.199
Keywords
This article is cited by
-
Chromosomes in the genomic age. Preserving cytogenomic competence of diagnostic genome laboratories
European Journal of Human Genetics (2021)
-
A Chinese multicenter retrospective study of isolated increased nuchal translucency associated chromosome anomaly and prenatal diagnostic suggestions
Scientific Reports (2021)
-
Whole genome analysis identifies the association of TP53 genomic deletions with lower survival in Stage III colorectal cancer
Scientific Reports (2020)
-
Recent Advances in the Noninvasive Prenatal Testing for Chromosomal Abnormalities Using Maternal Plasma DNA
Journal of Fetal Medicine (2020)
-
Low-pass genome sequencing: a validated method in clinical cytogenetics
Human Genetics (2020)
