
Abstract
The search for the genetic factors contributing to Alzheimer disease (AD) has evolved tremendously throughout the years. It started from the discovery of fully penetrant mutations in Amyloid precursor protein, Presenilin 1, and Presenilin 2 as a cause of autosomal dominant AD, the identification of the ɛ4 allele of Apolipoprotein E as a strong genetic risk factor for both early-onset and late-onset AD, and evolved to the more recent detection of at least 21 additional genetic risk loci for the genetically complex form of AD emerging from genome-wide association studies and massive parallel resequencing efforts. These advances in AD genetics are positioned in light of the current endeavor directing toward translational research and personalized treatment of AD. We discuss the current state of the art of AD genetics and address the implications and relevance of AD genetics in clinical diagnosis and risk prediction, distinguishing between monogenic and multifactorial AD. Furthermore, the potential and current limitations of molecular reclassification of AD to streamline clinical trials in drug development and biomarker studies are addressed.
Genet Med 18 5, 421–430.
Similar content being viewed by others

Main
Alzheimer disease (AD) is a devastating neurodegenerative disease and the predominant form of dementia (50–75%). In 2015, ~44 million people worldwide are estimated to have AD or a related dementia. Each year, 4.6 million new cases of dementia are predicted with numbers expected to almost double by 2030.1 AD is pathologically defined by severe neuronal loss, aggregation of amyloid β (Aβ) in extracellular senile plaques, and formation of intraneuronal neurofibrillary tangles consisting of hyperphosphorylated tau protein. The disease is clinically characterized by progressive deterioration of memory and cognitive functions, leading to loss of autonomy and ultimately requiring full-time medical care. Besides the strong impact of AD on the patient and primary caregivers, there is an enormous burden on society and public health due to the high costs associated with care and treatment of dementia. Aside from drugs that temporarily relieve symptoms, no treatment exists for AD.
Although the vast majority of patients develop clinical symptoms at age older than 65 years (late-onset AD), 2–10% of patients have an earlier onset of disease (early-onset AD). Rare autosomal dominant forms of AD exist, predominantly presenting as early-onset AD, although the majority of early-onset AD patients do not present with a clear autosomal pattern of inheritance. Nevertheless, the genetic predisposition of the non-Mendelian form of AD is considerable, even for late-onset AD patients, with a heritability estimate of 60–80%.2
The search for the genetic factors contributing to AD has evolved tremendously throughout the years, from the discovery of fully penetrant mutations in Amyloid precursor protein (APP), Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2) as a cause of autosomal dominant AD, and the identification of the ɛ4 allele of Apolipoprotein E (APOE) as strong genetic risk factor for both early-onset and late-onset AD one-quarter century ago, to the more recent identification of at least 21 additional genetic risk loci for the genetically complex form of AD in genome-wide association studies (GWAS) and massive parallel resequencing (MPS) efforts. Whereas the mutations in APP, PSEN1, and PSEN2 have been instrumental in the current understanding of the pathological process underlying AD, the findings from GWAS and MPS re-emphasize the multifactorial nature of AD. Nevertheless, translation of genetic findings into functional mechanisms that are biologically important in disease pathogenesis and treatment design remains a challenge. In this review, we position advances in AD genetics in light of the current endeavor toward translational research and personalized treatment. In the first part, we briefly discuss the current state of the art of AD genetics. In the second part, we review the potential and limitations of AD genetics in diagnosis and risk prediction, distinguishing between monogenic and multifactorial AD, and in molecular reclassification of AD to streamline clinical trials in drug development and biomarker studies.
Autosomal Dominant AD
With an estimated prevalence of <1%, autosomal dominant AD is very rare, but the discovery of pathogenic mutations in autosomal dominant AD pedigrees has brought about a breakthrough in the understanding of the pathogenesis of AD that reaches far beyond this small subgroup of AD. High-penetrant mutations in three genes were identified to cause autosomal dominant AD: APP,3,4 PSEN1,5,6,7 and PSEN2.8 Together, mutations in APP, PSEN1, and PSEN2 explain 5–10% of the occurrence of early-onset AD. The identification of mutations in these genes has not only provided important insights in the molecular mechanisms and pathways involved in AD pathogenesis but also led to valuable targets currently used in diagnosis and drug development.9
Amyloid precursor protein
APP is proteolytically processed by α-, β-, and γ-secretases following two pathways: the constitutive (nonamyloidogenic) or amyloidogenic pathway, leading to the production of different peptides. In the constitutive pathway, proteolysis of APP by α- and γ-secretases results in nonpathogenic fragments (sAPPα and α-C-terminal fragment). However, in the amyloidogenic pathway, enriched in neurons, the subsequent proteolysis of APP by β-secretase and γ-secretase gives rise to a mixture of Aβ peptides with different lengths. There are two major Aβ species: Aβ1–40 (90%) and Aβ1–42 (10%). The Aβ1–42 fragments are more aggregation-prone and are predominantly present in amyloid plaques in brains of AD patients. Of note, N- and C-truncated Aβ peptides also exist. A total of 39 APP mutations in 93 families are described, all of which affect proteolysis of APP in favor of Aβ1–42 (http://www.molgen.ua.ac.be/ADMutations/)10 (Supplementary Table S1 online), although some mutations also appear to affect N- or C-truncated peptides.3,4 In addition, APP duplications have been identified in autosomal dominant early-onset families.11,12 In the Icelandic population a rare protective variant in APP (p.A673T) has been observed, located near the β-proteolytic cleavage site and resulting in an impaired cleavage of APP, a reduction of Aβ1–40 and Aβ1–42 in vitro, and reduced AD risk.13 Interestingly, another mutation at the same position has been described (p.A673V) as pathogenic in the homozygous state but protective in the heterozygous state, suggesting a mixture of wild-type and mutant APP affects the aggregation properties of Aβ peptides.14
Presenilin 1 and presenilin 2
PSEN1 and PSEN2 are highly homologous genes. Mutations in PSEN1 are the most frequent cause of autosomal dominant AD known to date, whereas PSEN2 mutations are least frequent (Supplementary Table S1 online).5,6,7,8,15 Both proteins are essential components of the γ-secretase complex, which catalyzes the cleavage of membrane proteins, including APP. Mutations in PSEN1 and PSEN2 impair the γ-secretase mediated cleavage of APP in Aβ fragments, resulting in an increased ratio of Aβ1–42 to Aβ1–40, either through an increased Aβ1–42 production or decreased Aβ1–40 production, or a combination of both.16
PSEN1 mutations cause the most severe forms of AD with complete penetrance, and the onset of disease can occur as early as 25 years of age.10 The PSEN1 mutations have a wide variability of onset age (25–65 years), rate of progression, and disease severity. Missense mutations in the PSEN2 gene may show incomplete penetrance.8 In comparison to PSEN1 mutations, PSEN2 mutation carriers show an older age of onset of disease (39–83 years), but the onset age is highly variable among PSEN2-affected family members.5,8,17
Late-Onset AD and Genetic Risk
Late-onset AD is considered to be multifactorial; however, it involves a strong genetic predisposition.2 The genetic component itself is complex and heterogeneous, because there is no single model that explains the mode of disease transmission and gene mutations or polymorphisms may interact with each other and with environmental factors. For many years, APOE was the only major gene known to increase disease risk.18,19
Apolipoprotein E
APOE encodes a polymorphic glycoprotein expressed in liver, brain, macrophages, and monocytes. ApoE participates in transport of cholesterol and other lipids and is involved in neuronal growth, repair response to tissue injury, nerve regeneration, immunoregulation, and activation of lipolytic enzymes. The APOE gene contains three major allelic variants at a single gene locus (ɛ2, ɛ3, and ɛ4), encoding for different isoforms (ApoE2, ApoE3, and ApoE4) that differ in two sites of the amino acid sequence.19 The APOE ɛ4 allele increases risk in familial and sporadic early-onset and late-onset AD, but it is not sufficient to cause disease.18,19,20 The risk effect is estimated to be threefold for heterozygous carriers (APOE ɛ34) and 15-fold for ɛ4 homozygous carriers (APOE ɛ44), and has a dose-dependent effect on onset age.18,19 The APOE ɛ2 allele is thought to have a protective effect (OR = 0.6) and to delay onset age.20,21 Only 20–25% of the general population carries one or more ɛ4 alleles, where 40–65% of AD patients are ɛ4 carriers. ApoE binds to Aβ and effectuates the clearance of soluble Aβ and Aβ aggregations, and ApoE ɛ4 is thought to be less efficient in mediating Aβ clearance.22 The effect of APOE ɛ4 accounts for 27.3% of the estimated disease heritability of 80%.23 The part of the heritability that was yet unaccounted for has been the driving force behind decades of continued search for genetic risk factors.
Genome-wide association studies
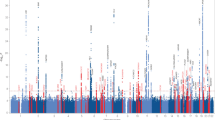
Large-scale collaborative GWAS and the International Genomics of Alzheimer’s Project have significantly advanced the knowledge regarding the genetic underpinnings of late-onset AD by identifying at least 20 additional genetic risk loci ( Table 1 ).23,24,25,26,27,28
None of these risk loci has an effect on AD risk of the magnitude of APOE ɛ4. Effect estimates range from an OR of 1.1–2.0 per risk allele. In contrast to APOE ɛ4, the population-attributable fraction of the individual genetic risk loci therefore remains limited, and their cumulative population-attributable fraction may not exceed the population-attributable fraction of APOE by much ( Table 1 ).29 The real merit of GWAS in AD genetics is probably that these studies have shed light on the pathophysiological pathways involved in AD ( Table 2 ). Although numerous GWAS-identified risk genes could be linked with the Aβ cascade and/or tau pathology, it was striking to note that the associated genes roughly cluster within three pathways: cholesterol and lipid metabolism; immune system and inflammatory response; and endosomal vesicle cycling.
Of note, the actual risk variants represented by these GWAS associations remain largely unidentified. This has implications for the translation of these findings into mechanistic insight, but also for obtaining accurate epidemiological estimates like population-attributable fraction and personal-level risk prediction (discussed in the next section). Moreover, several susceptibility loci are localized in gene-dense regions ( Table 3 ), and it remains to be determined which gene in these regions is responsible for the association.
The GWAS have sparked a wealth of genotype-phenotype correlation studies, most of which use GWAS proxy single-nucleotide polymorphisms rather than an underlying real risk variant. Efforts to identify the culprit variants at each locus are still limited, with most progress made in the genes identified in the first wave of sufficiently powerful GWAS.
Clusterin (CLU) was the first novel risk gene for AD identified simultaneously by two independent GWAS.24,25 CLU is a pleiotropic chaperone molecule that might be involved in AD pathogenesis through lipid transport, inflammation, and directly by influencing Aβ aggregation and clearance from the brain by endocytosis. A GWAS-identified single-nucleotide polymorphism has been proposed as a functional variant directly affecting alternative splicing of CLU,30 and targeted resequencing of CLU led to the detection of an increased frequency of rare coding CLU variations in patients, independent of the common association signal identified in GWAS, suggesting that different variants (rare coding and common regulatory) within a single locus can exist and have independent effects on the disease.31
Similar observations have been made more recently for Sortilin-related receptor-1 (SORL1) and ATP-binding cassette subfamily A member 7 (ABCA7), although for both genes the rare variants seem to have a higher penetrance than the rare variants observed in CLU.
SORL1 was identified as a risk factor for late-onset AD through a candidate gene approach,32 and findings were confirmed in the recent International Genomics of Alzheimer’s Project GWAS meta-analysis.23 Although the functional variants underlying these association signals remain unclear, exploring the effect of a common risk haplotype at SORL1 in human induced pluripotent stem cells suggests that these genetic variants might act as effect modifiers of the induction of SORL1 expression and APP processing by BDNF.33 However, whole-exome sequencing revealed several SORL1 nonsense and missense mutations in patients with autosomal dominant early-onset AD34 and targeted resequencing in late-onset AD patients revealed additional missense mutations in SORL1, including a common variant, which segregated with disease and affected APP processing in vitro,35 indicating that the mutation spectrum of SORL1 in AD pathogenesis contains both common and rare variants. ABCA7 was first identified as a risk gene for AD in the GWAS setting28 and is highly expressed in hippocampal neurons, one of the earliest affected brain regions of AD patients, and in microglia. It remains unclear whether ABCA7 influences AD risk via interacting with APOE and lipid metabolism, functioning as an immune system molecule and clearing of Aβ aggregates from the brain or a combination of both. Several studies have reported an association between AD risk variants and ABCA7 expression in brain,36 albeit with discrepant findings. More recently, an increased frequency of rare loss-of-function mutations in ABCA7 has been described in AD patients,37 which may present with an autosomal dominant pattern of inheritance.38
Bridging integrator 1 (BIN1) is involved in clathrin-mediated endocytosis, a process essential for the recycling of synaptic vesicles after each synaptic release.24,26 BIN1 expression levels are increased in human brain and are associated with later disease onset and shorter disease duration in AD patients.39,40 A functional follow-up study identified a 3 bp insertion upstream of the BIN1 gene as a putative functional candidate, increasing transcriptional activity in vitro, BIN1 expression levels in human brain, and AD risk. Further, increased P-tau181P has been described in cerebrospinal fluid of AD patients, and the Drosophila ortholog Amph modifies tau neurotoxicity, hypothesizing that BIN1 mediates AD risk through Tau pathology.40
Complement component (3b/4b) receptor 1 (CR1) has multiple functions, including the regulation of complement activation and functions as a mediator of the innate immunity. CR1 has the capacity to bind complement components C3b and C4b and is expressed in many cell types in particular cells of the circulatory system.41 An intragenic functional copy-number variation in CR1 has been proposed as the variant underlying the GWAS association between CR1 and AD risk. This copy-number variation translates into two major isoforms, CR1-F and CR1-S, resulting in a variable number of C3b/C4b and cofactor activity binding sites at the receptor, which are important in the complement cascade.41,42 Although the exact mechanism of action remains unclear, first explorations in brain samples suggest that the CR1-S isoform is expressed at lower protein levels than CR1-F, and therefore is probably linked to increased complement activation. Both isoforms show a different pattern of CR1 distribution in neurons, which could indicate that CR1-S and CR1-F isoforms are differentially processed in neurons.42
CD33 is an immune cell surface receptor promoting cell-cell interactions and regulating cell functions in the innate and adaptive immune systems through clathrin-mediated endocytosis.27,28 CD33 is expressed on myeloid cells and microglia, and expression is specifically increased in brain microglial cells, which correlated with amyloid plaque burden and advanced cognitive decline.39 Microglial cells expressing CD33 show impaired Aβ phagocytosis and are correlated with amyloid plaque burden in AD brains.43 A variant, putatively involved in alternative splicing, has been proposed as a functional variant.44
Massive parallel resequencing
Through advances in MPS technologies different research groups have successfully identified low frequency and rare variants. Multiple rare, missense variants in Triggering Receptor Expressed On Myeloid Cells 2 (TREM2) have been reported to increase risk for late-onset AD.45,46 The most common variant in European populations R47H was reported to increase AD risk threefold. The observed effect size of R47H is comparable to the effect of the APOE ɛ4 allele; the frequency of the variant, however, is low (MAF ~0.3%), meaning the impact on population level of the TREM2 variant is much lower. These findings were replicated in different populations of European origin, but not in Asian subjects, demonstrating that certain risk variants may be population-specific or that the same gene can harbor different disease-associated variants.
A family-based study design led to the detection of rare variants within the Phospholipase D3 (PLD3) gene associated with AD risk. Whole-exome sequencing identified variant V232M, which segregates with disease status in late-onset AD families47 and is associated with a two- to threefold increase in disease risk. However, several independent studies were unable to replicate this observation.48 Numerous large-scale MPS studies on AD are ongoing and are anticipated to bring about the next wave of gene discovery for AD.
Clinical Implications and Relevance of Genetic Findings
Genetics in diagnosis and risk prediction of autosomal dominant AD
Although our knowledge of the genetic causes and risk factors of AD is advancing, the question arises how to translate and implement these insights into improved public health. The most direct implementation, which is already available to patients and relatives today, is genetic diagnostic and predictive screening for causal mutations in APP, PSEN1, and PSEN2. These causal mutations, however, are only responsible for a small portion of AD patients. For a significant number of patients for whom genetic diagnostic screening is requested, the tests will therefore be negative without excluding a genetic cause of disease. For instance, in our Diagnostic Service Facility (http://www.molgen.ua.ac.be/DNADiagnostics/), pathogenic mutations are identified in only 4, 1.4, and 1% of patients referred for diagnostic screening for PSEN1, APP, and PSEN2, respectively, despite prior evidence for a monogenic cause of AD. Nevertheless, the ability to identify an explanation for the clustering of AD in a family and the ability to use this toward predictive testing in subsequent generations provide an important step toward autonomy of patients and at-risk individuals. Comprehensive genetic counseling protocols are available for AD diagnostic and predictive testing to provide a framework for clinicians and geneticists to evaluate which patients may benefit from genetic testing.49 Important ethical considerations should be taken into account, including the disclosure of AD diagnosis and genetic test results, the social stigma, employment, and family planning.50 Moreover, the identification of a mutation is not a certain predictor of disease or onset age, given that these mutations can vary in terms of penetrance and gene expression.10,51 Several studies have attempted to discover onset age modifiers. A dose-dependent effect of APOE ɛ4 on onset age has been described, showing a lower mean onset age in AD patients for each additional copy of the ɛ4 risk allele. The mean onset age decreased from 84 to 68 years.19 The effect of APOE ɛ4 on onset age of autosomal dominant AD was confirmed in large pedigrees.52 A review of demographic data on 387 autosomal dominant AD families, however, suggests that factors like parental onset age, mean onset age of family members, and mean onset age of carriers with the same mutation may be stronger predictors of onset age in a mutation carrier than APOE ɛ4.51 In a Columbian kindred carrying the PSEN1 p.E280A mutation, a protective haplotype was recently identified through whole genome sequencing. This haplotype, associated with a 10-year delay in onset age, harbors a missense mutation in the CCL11 gene encoding eotaxin-1.53 Evidence exists for additional onset age modifiers detected through linkage analyses; however, the specific genes driving these linkage signals remain unknown.54 Gene–environment interactions and epigenetic changes can also result in significantly different disease outcomes.55
With the advent of high-capacity MPS, genetic diagnostic testing is entering a new era. Multiple genes can now be screened simultaneously using disease-oriented or disease spectrum–oriented gene panels, obviating the need of decision trees based on clinical parameters. For AD, a disease-spectrum approach may be particularly relevant. For example, the mutation p.R406W in MAPT, a known causal gene for FTLD, has repeatedly been reported in pedigrees with a clinical presentation of AD.56 Mutations in two other FTLD genes, GRN and C9orf72, have also been described in clinical AD cohorts.57,58 It may be important to include screening of these genes in the genetic diagnostic work-up of high genetic load AD patients, particularly in light of the fact that APP, PSEN1, and PSEN2 account for only a small proportion of autosomal dominant AD.
These advancing techniques could represent fast and cost-effective tools in a clinical setting, but with the incorporation of new technologies, both a greater complexity in the interpretation of genetic results and additional ethical issues arise. For instance, prior to the availability of MPS, mutation screening of APP focused only on the exons encoding the Aβ peptide and its cleavage sites. Now that screening of the full locus has become highly feasible, this may reveal variants of unknown significance. This complicates the interpretation and impact of the information on patients and society. An additional challenge will be the prospect of secondary or incidental findings, particularly when whole exome or genome sequencing is performed rather than gene panel-based sequencing. These findings might be relevant for patient management but not related to the phenotype. Incidental findings have great implications concerning the return of genomic information to patients or research participants. This necessitates extensive guidelines and genetic counseling.59 A recent study investigated the opinion and expectations of stakeholders concerning an opportunistic screening of data sets by researchers and the return of incidental but health-related findings to the patient. The results indicated that 88% of the stakeholders thought that incidental findings should be made available to research participants. Despite the interest in access to data results, 69% of the responders did not expect researchers to actively search for incidental findings not relevant to their research in exome or genome data sets.60
Genetic risk prediction in complex AD
The role of genetics in diagnosis and risk prediction in late-onset complex AD is much less straightforward. Despite the established evidence of APOE ɛ4 as a risk factor for AD, its value in disease prediction in a clinical setting is limited, not only due to the restriction of current therapeutic consequences but also because APOE ɛ4 is neither necessary nor sufficient to cause the disease. Up to 75% of individuals heterogeneous for APOE ɛ4 do not develop AD during life, and up to 50% of people with AD do not carry the high-risk ɛ4 allele.20
The relevance of clinical testing for common genetic variations identified in GWAS is even more limited, because these variations confer smaller relative risks and collectively explain only a small proportion of the underlying genetic contribution. Moreover, genetic testing would only provide an assumption of an individual’s risk of developing AD since these variants represent indirect markers of association and not the true disease-related functional variants. Combining multiple susceptibility loci into a global genetic risk score (GRS) might improve the prediction of individuals at risk. For example, an 8-single-nucleotide polymorphism GRS was associated with an accelerated progression from mild cognitive impairment to AD.61 Another study reported that a GRS including nine non-APOE alleles in a model allowing for interaction between loci significantly improved the ability to predict AD compared to APOE alone; however, the improvement did not reach the sensitivity or specificity necessary for clinical diagnostic use.29 We investigated a GRS combining 22 AD susceptibility loci. The best model was a weighted GRS that took into account the different risk effects of APOE in different age groups.62,63 This model performed significantly better than APOE alone in discriminating AD patients from healthy elderly. Despite the fact that this model incorporated all reliable GWAS association signals reported thus far, as well as the established rare risk variant in TREM2, the model only achieved a sensitivity of 55% and a specificity of 78%, impeding use in clinical practice.63 More gain in predictive ability is to be expected from inclusion of true functional variants rather than GWAS signals, from incorporation of epistatic effects,29 or combination with nongenetic biomarkers.
Impact of genetic testing on individuals
Genetic and susceptibility testing brings concerns regarding the impact of test results on individuals. Both survey data and clinical research have shown that the majority of individuals at risk for AD are interested in knowing their genetic profile.64 Furthermore, since the emergence of personal genomics companies offering direct-to-consumer testing for various neurodegenerative diseases, it is of great importance to examine the possible implications of these test results. Direct-to-consumer companies do not provide genetic counseling or exclude psychologically vulnerable consumers, which increases the potential for inadequately understanding the meaning or implications of test results.55
A series of clinical studies, part of the REVEAL trial, has been conducted to investigate the psychological and behavioral impact of genetic screening results. In the REVEAL study, risk estimates for AD were established based on age, sex, family history, and APOE genotype. No evidence of fatalism was observed among participants receiving APOE ɛ4–positive results; however, modest evidence of false reassurance was detected among participants receiving APOE ɛ4–negative results. Further, APOE testing in at-risk individuals with a positive family history does not pose significant psychological risks like depression or anxiety when they are provided by proper pre- and posttest counseling. A multisite clinical trial evaluated the impact of susceptibility testing with APOE and further assessed distress of deterministic genetic testing by disclosing PSEN1 or PSEN2 information to individuals at risk for AD. This study suggested that the test-related distress experienced by those receiving positive results for a deterministic mutation is similar to the distress experienced by those receiving positive results from genetic susceptibility. The most common behavioral change reported in the REVEAL study was the use of nutritional supplements, suggesting that genetic susceptibility testing may enhance the preference for biological interventions like medication over health behavior changes like a lifestyle change.55
Drug development and clinical trials
Development of disease-modifying and symptomatic therapeutics for AD to date has mostly focused on early insights on the molecular mechanisms and pathways involved in AD and specifically followed three AD hypotheses: the cholinergic, amyloid cascade, and tau hypotheses.9 Therapeutics based on the enhancement of the cholinergic system show consistent, but modest, clinical effects in late-phase trials.9 A substantial portion of the field focused its efforts on the amyloid cascade hypothesis, highlighted by the identification of pathogenic mutations in APP, PSEN1, and PSEN2. This approach led to human clinical trials potentially decreasing the production or aggregation of Aβ or enhancing Aβ clearance from the brain. Recent passive and active anti-tau immunization studies in mouse models have been proven effective at preventing and improving tau pathology.65 The progression of neurofibrillary tangles pathology throughout the brain correlates strongly with synaptic and neuronal loss and cognitive decline, and makes it a potential therapeutic target to interrupt progression of tau pathology early in disease. The spread of tau pathology and neuronal tau release is thought to be a regulated process through active secretion and interneuronal transfer of tau, which could facilitate transneuronal spread of tangle pathology.66
Despite considerable advances in the knowledge of AD pathogenesis, the AD field has struggled to move druggable targets from preclinical research into effective therapies. Although there may be numerous pharmacological reasons for this, it might be partly explained by the study design and patient recruitment in phase III clinical trials. Evidence accumulates that AD pathology is progressing silently for decades before the appearance of clinical symptoms, and neuronal loss is already present at the stage of mild cognitive impairment.67 Treating symptomatic patients with full-blown disease may not be effective, and therapeutics applied earlier in the course of AD would be more likely to achieve effective disease modification. This concept has been supported by several preclinical research successes in AD mouse models.68 Although the prospect for preventing neurodegenerative AD processes seems challenging at the moment, this approach is well-accepted and successful in other medical fields, like cardiovascular disease.69
Moreover, the majority of these trials have enrolled patients with a clinical diagnosis of late-onset AD. The success rate of therapeutic and biomarker trials might considerably be improved if trial design would foresee preselection of study participants, representing an etiologically more homogeneous group that has the highest chance of benefiting from specific treatments and fewer chance of showing adverse effects.
Genetic testing can play an important role in this endeavor. To investigate early disease changes, patients cohorts such as the Dominantly Inherited Alzheimer’s Network can be specifically selected for mutation carriers of APP, PSEN1, and PSEN2.70 Furthermore, clinically normal individuals thought to be on the trajectory toward the symptomatic stages of AD can be detected through advances in genetic testing for APOE or more comprehensive genetic risk scores alongside the more generally accepted biomarkers like amyloid and tau in cerebrospinal fluid and PET neuroimaging.67
Several clinical trials have been initiated that focus on presymptomatic or early symptomatic, more homogenous study cohorts on the basis of their genetic profile. For instance, a phase II/III clinical trial of two drugs is ongoing for preclinical carriers of mutations in APP, PSEN1, and PSEN2 in the context of the Alzheimer Prevention Initiative (ClinicalTrials.gov). The TOMMORROW trial is a phase III trial examining the effect and ability of pioglitazone, a peroxisome proliferator–activated receptor γ agonist, to delay the onset of mild cognitive impairment of AD in individuals at high risk. Risk prediction is based on an algorithm including APOE and TOMM40 genotypes. A poly-T repeat in TOMM40 has been described to increase risk of AD.71 However, TOMM40 is in very strong linkage disequilibrium with APOE; therefore, the independent role of TOMM40 as a risk gene is still under discussion. This trial may give further insight regarding the role of TOMM40 in AD risk.72 A phase II trial of the Aβ-specific antibody bapineuzumab stratified individuals based on APOE ɛ4 status. Analysis suggested that APOE ɛ4–negative subjects had a better response compared with placebo than the APOE ɛ4–positive study participants, who showed worse Aβ pathology and an earlier onset of symptoms.73
Although no clinical trials are yet incorporating full genetic risk profiles into their design, genetic risk profiles are a promising tool in genetic classification. A high GRS was found predictive of positive family history, younger disease onset, and lower cerebrospinal fluid Aβ1–42 levels,63 implying that a genetic preselection may be a fast and cost-efficient way to identify individuals at increased risk for AD for clinical or biomarker trials, prior to more cost-intensive and labor-intensive methods like amyloid imaging. In addition, molecular subclassification will be possible by determining pathways enriched for risk alleles within an individual, at least to the extent to which associated single-nucleotide polymorphisms can currently be assigned to single genes within a locus. This will be particularly relevant when testing therapeutics targeting a specific pathway.63,74
More complex risk profiling by integrating additional nongenetic information concerning AD endophenotypes, such as cerebrospinal fluid biomarkers and neurofibrillary pathology, into a genetic risk profile may aid in the selection of individuals at high risk and the prediction of future cognitive decline.75 As with personal genetic and susceptibility testing, the identification of high risk individuals for research purposes has important ethical implications, which requires careful attention.
Conclusion
Shifting research toward genetic molecular profiling using integrated -omics approaches has led to considerable progress in complex diseases such as those addressed by the cancer research field. Advances in genetics and clinically relevant NGS applications including whole-genome sequencing, whole-exome sequencing, transcriptome profiling, as well as epigenomic and proteomic characterization have paved the way to molecular profiling of cancer subtypes and provide important instructions for other complex diseases like AD. Although there is still a long way to go in the AD field before precision medicine is achieved, there is reason for cautious optimism with the continued elucidation of novel genes involved in AD and the impact that genetic profiling can have can shift toward prediction and prevention.
Disclosure
The authors declare no conflict of interest.
References
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 2013;9:63–75.e2.
Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174.
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991;349:704–706.
St George-Hyslop PH, Tanzi RE, Polinsky RJ, et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987;235:885–890.
Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995;375:754–760.
St George-Hyslop P, Haines J, Rogaev E, et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet 1992;2:330–334.
Van Broeckhoven C, Backhovens H, Cruts M, et al. Mapping of a gene predisposing to early-onset Alzheimer’s disease to chromosome 14q24.3. Nat Genet 1992;2:335–339.
Sherrington R, Froelich S, Sorbi S, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet 1996;5:985–988.
Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med 2014;275:251–283.
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 2012;33:1340–1344.
Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 2006;38:24–26.
Sleegers K, Brouwers N, Gijselinck I, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006;129(Pt 11):2977–2983.
Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012;488:96–99.
Di Fede G, Catania M, Morbin M, et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009;323:1473–1477.
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995;269:973–977.
Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer’s disease. Hum Mutat 1998;11:183–190.
Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010;133(Pt 4):1143–1154.
Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993;43:1467–1472.
Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921–923.
Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356.
Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–184.
Deane R, Sagare A, Hamm K, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 2008;118:4002–4013.
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al.; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45:1452–1458.
Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009;41:1088–1093.
Lambert JC, Heath S, Even G, et al.; European Alzheimer’s Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009;41:1094–1099.
Seshadri S, Fitzpatrick AL, Ikram MA, et al.; CHARGE Consortium; GERAD1 Consortium; EADI1 Consortium. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840.
Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436–441.
Hollingworth P, Harold D, Sims R, et al.; Alzheimer’s Disease Neuroimaging Initiative; CHARGE consortium; EADI1 consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011;43:429–435.
Ebbert MT, Ridge PG, Wilson AR, et al. Population-based analysis of Alzheimer’s disease risk alleles implicates genetic interactions. Biol Psychiatry 2014;75:732–737.
Szymanski M, Wang R, Bassett SS, Avramopoulos D. Alzheimer’s risk variants in the clusterin gene are associated with alternative splicing. Transl Psychiatry 2011;1:e18.
Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener 2012;7:3.
Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 2007;39:168–177.
Young JE, Boulanger-Weill J, Williams DA, et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 2015;16:373–385.
Pottier C, Hannequin D, Coutant S, et al.; PHRC GMAJ Collaborators. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 2012;17:875–879.
Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015;77:215–227.
Vasquez JB, Fardo DW, Estus S. ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci Lett 2013;556:58–62.
Steinberg S, Stefansson H, Jonsson T, et al.; DemGene. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 2015;47:445–447.
Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol 2015;14:814–822.
Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One 2012;7:e50976.
Chapuis J, Hansmannel F, Gistelinck M, et al.; GERAD consortium. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 2013;18:1225–1234.
Brouwers N, Van Cauwenberghe C, Engelborghs S, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry 2012;17:223–233.
Hazrati LN, Van Cauwenberghe C, Brooks PL, et al. Genetic association of CR1 with Alzheimer’s disease: a tentative disease mechanism. Neurobiol Aging 2012;33:2949.e5–2949.e12.
Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013;78:631–643.
Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 2013;33:13320–13325.
Guerreiro R, Wojtas A, Bras J, et al.; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117–127.
Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 2013;368:107–116.
Cruchaga C, Karch CM, Jin SC, et al.; UK Brain Expression Consortium; Alzheimer’s Research UK Consortium. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014;505:550–554.
Lambert JC, Grenier-Boley B, Bellenguez C, et al. PLD3 and sporadic Alzheimer’s disease risk. Nature 2015;520:E1.
Goldman JS, Hahn SE, Catania JW, et al.; American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med 2011;13:597–605.
Gauthier S, Leuzy A, Racine E, Rosa-Neto P. Diagnosis and management of Alzheimer’s disease: past, present and future ethical issues. Prog Neurobiol 2013;110:102–113.
Ryman DC, Acosta-Baena N, Aisen PS, et al.; Dominantly Inherited Alzheimer Network. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014;83:253–260.
Pastor P, Roe CM, Villegas A, et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol 2003;54:163–169.
Kosik KS, Lalli M, Arcila M, et al. Unravelling the genetics of a large familial AD kindred in Antioquia Colombia. Neurodegener Dis 2015;15:159–288.
Marchani EE, Bird TD, Steinbart EJ, et al. Evidence for three loci modifying age-at-onset of Alzheimer’s disease in early-onset PSEN2 families. Am J Med Genet B Neuropsychiatr Genet 2010;153B:1031–1041.
Roberts JS, Uhlmann WR. Genetic susceptibility testing for neurodegenerative diseases: ethical and practice issues. Prog Neurobiol 2013;110:89–101.
Rademakers R, Dermaut B, Peeters K, et al. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum Mutat 2003;22:409–411.
Brouwers N, Nuytemans K, van der Zee J, et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 2007;64:1436–1446.
Cacace R, Van Cauwenberghe C, Bettens K, et al. C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 2013;34:1712.e1–1712.e7.
Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet 2013;14:295–300.
Middleton A, Morley KI, Bragin E, et al.; Deciphering Developmental Disorders Study. No expectation to share incidental findings in genomic research. Lancet 2015;385:1289–1290.
Rodríguez-Rodríguez E, Sánchez-Juan P, Vázquez-Higuera JL, et al. Genetic risk score predicting accelerated progression from mild cognitive impairment to Alzheimer’s disease. J Neural Transm 2013;120:807–812.
Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 2011;16:903–907.
Sleegers K, Bettens K, De Roeck A, et al.; BELNEU consortium. A 22-single nucleotide polymorphism Alzheimer risk score correlates with family history, onset age, and cerebrospinal fluid Aβ42. Alzheimers Dement; e-pub ahead of print 15 June 2015.
Cassidy MR, Roberts JS, Bird TD, et al. Comparing test-specific distress of susceptibility versus deterministic genetic testing for Alzheimer’s disease. Alzheimers Dement 2008;4:406–413.
Pooler AM, Polydoro M, Wegmann S, Nicholls SB, Spires-Jones TL, Hyman BT. Propagation of tau pathology in Alzheimer’s disease: identification of novel therapeutic targets. Alzheimers Res Ther 2013;5:49.
de Calignon A, Polydoro M, Suárez-Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012;73:685–697.
Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 2014;84:608–622.
Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004;43:321–332.
Leitersdorf E, Muratti EN, Eliav O, Peters TK. Efficacy and safety of triple therapy (fluvastatin-bezafibrate-cholestyramine) for severe familial hypercholesterolemia. Am J Cardiol 1995;76:84A–88A.
Fagan AM, Xiong C, Jasielec MS, et al.; Dominantly Inherited Alzheimer Network. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med 2014;6:226ra30.
Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 2010;10:375–384.
Roses AD, Saunders AM, Lutz MW, et al. New applications of disease genetics and pharmacogenetics to drug development. Curr Opin Pharmacol 2014;14:81–89.
Salloway S, Sperling R, Gilman S, et al.; Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009;73:2061–2070.
Kohannim O, Hua X, Rajagopalan P, et al.; Alzheimer’s Disease Neuroimaging Initiative. Multilocus genetic profiling to empower drug trials and predict brain atrophy. Neuroimage Clin 2013;2:827–835.
Martiskainen H, Helisalmi S, Viswanathan J, et al. Effects of Alzheimer’s disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. J Alzheimers Dis 2015;43:565–573.
Acknowledgements
Research by the authors’ group is funded in part by the Belgian Science Policy Office Interuniversity Attraction Poles Program; the Alzheimer Research Foundation (SAO-FRA); the Flemish government–initiated Excellence Program Methusalem (EWI); the Flemish government–initiated Flanders Impulse Program on Networks for Dementia Research (VIND); the Research Foundation Flanders (FWO); the Agency for Innovation by Science and Technology (IWT); the University of Antwerp Research Fund (http://www.uantwerpen.be/); the European Commission FP7 Seventh Framework Programme for research, technological development, and demonstration under grant agreement 305299 (AgedBrainSYSBIO); and the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement 115372, the resources of which are composed of financial contributions from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in-kind contribution.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 41 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18, 421–430 (2016). https://doi.org/10.1038/gim.2015.117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.117
Keywords
This article is cited by
-
Alzheimer’s polygenic risk scores, APOE, Alzheimer’s disease risk, and dementia-related blood biomarker levels in a population-based cohort study followed over 17 years
Alzheimer's Research & Therapy (2023)
-
Novel Alzheimer’s disease genes and epistasis identified using machine learning GWAS platform
Scientific Reports (2023)
-
Investigating molecular interactions between human transferrin and resveratrol through a unified experimental and computational approach: Role of natural compounds in Alzheimer’s disease therapeutics
Amino Acids (2023)
-
Advances in protein glycosylation and its role in tissue repair and regeneration
Glycoconjugate Journal (2023)
-
Complexity decline of hippocampal CA1 circuit model due to cholinergic deficiency associated with Alzheimer’s disease
Cognitive Neurodynamics (2023)
