
Abstract
Purpose:
Technological advances now allow for multiplex platforms to simultaneously test many genetic conditions. Typically, such platforms are validated by assaying samples with known genotypes and/or phenotypes and/or with synthetic plasmids; however, these methods have limitations and with the inclusion of rarer diseases and mutations, we can no longer rely solely on them. We used a novel genomic database to validate an expanded genetic carrier screening platform.
Methods:
Our expanded carrier screening assay uses the Illumina Infinium iSelect HD Custom genotyping platform to test for 213 genetic diseases by assaying 1,663 pathogenic mutations. We leveraged two Coriell Institute biorepositories for validation: the Subcollection of Heritable Diseases and the 1000 Genomes Project.
Results:
We measured 12,394 mutation observations in 206 samples, resulting in 246 true positives, 12,147 true negatives, 1 false positive, and no false negatives. Results demonstrated high sensitivity (99.99%) and specificity (99.99%).
Conclusion:
We successfully validated our platform with two biorepositories, demonstrating high sensitivity and specificity. The 1000 Genomes Project samples provided both positive and negative validation for mutations in genes not available through other biorepositories, expanding the depth of validated variants. We recommend including samples from the 1000 Genomes Project in the validation of future multiplex testing platforms.
Genet Med advance online publication 30 July 2015
Similar content being viewed by others

Main
There are over 6,000 known single-gene disorders, which are present in ~1/300 of the 4,000,000 babies born in the United States each year and account for at least 10% of pediatric hospitalizations and 20% of infant deaths.1,2,3,4 Traditionally, genetic carrier screening is performed for only a limited number of single-gene disorders, based on an individual’s ethnicity, family history, and partner’s carrier status.5 However, screening based on ethnicity alone has significant limitations. The genetic pool is homogenizing, and people are becoming less aware of—and less likely to identify with—a specific ethnicity.6 Moreover, recent advances in genotyping and sequencing technologies now allow simultaneous testing to be performed for a larger number of diseases and mutations, at a significantly lower cost than previous methods. Several professional societies have recognized the rationale for expanded, panethnic screening and have published position statements regarding the incorporation of such platforms into clinical practice for patients during pregnancy or before conception.5
Multiplex genotyping platforms have been validated using a number of different methods. A common method of validation is to assay samples with a known genotype and/or phenotype sourced from biorepositories, such as the Coriell Subcollection of Heritable Diseases.7,8 However, these biorepositories provide a limited number of available validation samples for these genotyping assays. Another method that has been implemented is the use of plasmids containing assay-specific mutations.7 However, this method is also limited; for example, the gene-specific sequence included in the plasmid is not representative of full-length genomic DNA and may therefore affect the ability of the assay to detect the mutation being targeted.9 Additionally, the mixture of plasmids and genomic DNA can interfere with accurate mutation detection.10 These current validation methods are not sufficient to test the analytical validity of multiplex genotyping platforms screening for an increasing number of rare diseases and mutations, which are now routinely used in a clinical setting.
To address the limitations of current validation methods, we used the 1000 Genomes Project, a global catalog of genetic variation across numerous ethnic populations using high-throughput sequencing.11,12 Unlike most biorepositories, samples from the 1000 Genomes Project have curated sequencing results, allowing for validation of multiple variants, including true positives and true negatives, within every sample. The accessibility of these data addresses the issues and limitations of previous validation methods, representing a novel approach for validation of a multiplex genotyping platform.
We designed a custom, panethnic, expanded genetic carrier screening assay built on the Illumina Infinium iSelect HD Custom genotyping platform. Our customized multiplex array screens for 213 genetic diseases by testing for 1,663 pathogenic mutations. By using the 1000 Genomes Project in combination with a more traditional biorepository, we successfully validated our multiplex genotyping platform to show high sensitivity and specificity.
Materials and Methods
Disease and mutation selection and curation
Diseases were selected for inclusion on the assay based on the following criteria: (i) autosomal recessive or X-linked recessive inheritance; (ii) disease phenotype that affects life expectancy and/or quality of life; (iii) inclusion in professional society recommendations (e.g., American Congress of Obstetricians and Gynecologists, American College of Medical Genetics and Genomics),5 newborn screening guidelines,13 disease advocacy group opinions (e.g., the Jewish Genetic Disease Consortium14); and (iv) preimplantation genetic diagnosis for the condition. A total of 213 autosomal recessive and X-linked diseases were selected for screening. An extensive literature search for population-wide studies and individual case study reports was conducted to select 1,663 associated pathogenic mutations, including point mutations, insertions, and deletions across intronic and exonic regions; splice-site mutations; and promoter region mutations.
Probe design
For each mutation, single-base extension probes were designed according to specifications outlined in Illumina’s custom design process for the Infinium iSelect HD Custom Genotyping BeadChip microarray. The Reference Sequence transcripts from the February 2009 release of hg19/Genome Reference Consortium (GRCh37) were used to design multiple probes for each mutation included in the assay.15 Probes were designed using flanking sequences up to 100 nucleic acids upstream and downstream of each mutation. Flanking regions were checked for degeneracy within 50 bases of each mutation. In the event that nonpathogenic polymorphisms existed within this region, additional probes were developed to account for the different possible flanking region combinations. Consider this example:
Primer 1: 3′ (C/A)AGATAATCA...CTAGCA 5′
Primer 2: 3′ (C/A)AGACAATCA...CTAGCA 5′
Here, a set of probes was developed targeting a C>A substitution. Four bases upstream of the point of interest exists a T>C polymorphism. To account for individuals with each genotype, probes were designed for both sequences.
Sample ascertainment
Samples were selected from two biorepositories housed by the Coriell Institute (Camden, NJ): the Subcollection of Heritable Diseases and the 1000 Genomes Project.
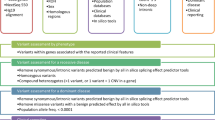
Validation samples of mutations included in our assay were selected from the Subcollection of Heritable Diseases based on known status as heterozygous, homozygous, or compound heterozygous. A total of 126 genomic DNA samples covering 94 unique mutations were used for validation, providing 161 true-positive observations (Supplementary Table S1 online).
A total of 80 samples from the 1000 Genomes Project were selected, with specific attention to samples from a diverse range of ethnic groups (Supplementary Table S2 online). For each ethnic group selected, at least one male and one female sample were included to cover mutations found on the sex chromosomes. We focused on validating 155 mutations in each selected sample, of which 49 mutations were reported as heterozygous within these samples. Multiple samples were heterozygous and/or wild-type for the same genetic variant; this provided 86 true-positive observations and 12,147 true-negative observations for sensitivity and specificity calculations (Supplementary Table S2 online).
Only 8 mutations overlapped between the samples ascertained from the two biorepositories, resulting in a total of 133 unique mutations. Of the 206 samples selected for validation, 106 were female and 100 were male.
Genotyping assay
All DNA samples were prepared and purified following the QIAamp DNA Purification Protocol via QIAcube (QIAGEN, Venlo, Limburg, The Netherlands). Samples were assayed using the Infinium iSelect HD Custom Genotyping BeadChip platform (Illumina, San Diego, CA). This is a two-channel assay in which fluorescently-labeled nucleotides (red and green) are used to genotype each mutation. All analyses were performed in a CLIA-certified laboratory (Reprogenetics, Livingston, NJ).
Bioinformatics analysis and quality control
The resulting genotype for each of the 1,663 genetic mutations assayed was analyzed through a clustering algorithm via the GenomeStudio Genotyping Module version 1.0 (Illumina). Genotypes are called based on the intensity of the two fluorescent signals for each assayed variant. These two intensities are translated into normalized values (x and y) for each probe, corresponding to a specific genetic variant. The normalized values are used to calculate R, the total allele intensity from both channels (x + y), and theta, the allelic intensity ratio (2/π*arctan(y/x)). Based on R and theta for each probe, a genotype for each mutation is reported in a standard homozygous wild-type, heterozygous, and homozygous mutant format.
Results
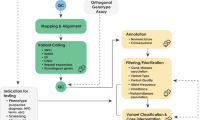
Validation results allowed us to effectively assess the accuracy, sensitivity, and specificity of our genotyping assay. A total of 206 samples sourced from the Coriell Subcollection of Heritable Diseases and 1000 Genomes Project were analyzed (Supplementary Tables S1 and S2 online). Samples varied in gender, ethnicity, and carrier/affected status to maximize the pool of variants for validation (Supplementary Tables S1 and S2 online). Genotype calls were reported as homozygous wild-type, heterozygous, or homozygous mutant according to the clustering algorithm as analyzed by Illumina’s GenomeStudio Module version 1.0 (Figure 1 ).

Example of a cluster graph used for calling genotypes: Tay Sachs c.1278_1279insTATC. Visual interpretation of genotype calls for multiple samples based on R and theta demonstrate how each genotype clusters based on allele intensity ratios. Each dot corresponds to a unique sample. Blue indicates homozygous wild-type; orange, heterozygous mutant; green, homozygous mutant. The homozygous mutant case in this example is a validation sample from the Coriell Subcollection of Heritable Diseases (NA11852).
Assay-generated genotype calls then were compared with the genotypes provided by the Coriell Subcollection of Heritable Diseases and the 1000 Genome Project database and demonstrated the high sensitivity (99.99%) and specificity (99.99%) of our genotyping assay (Table 1 ). We measured 12,394 mutation observations, resulting in 246 true positives, 12,147 true negatives, and no false negatives. The assay yielded one false-positive result for mutation c.1448T>C/p.L444P (rs421016) in the GBA gene associated with Gaucher disease.
Discussion
Through our validation, we have demonstrated the high sensitivity, specificity, negative predictive value (NPV), and positive predictive value (PPV) of our assay (Table 1 ). Not only have we shown the benefits of using the 1000 Genomes Project as a validation tool, but we have also provided the necessary analytical validity and support for the use of our platform as a clinical expanded carrier screen.
Traditional validation studies relied on samples with a known genotype and a corresponding single phenotype, such as those available through the Subcollection of Heritable Diseases, or used synthesized plasmids.7,8 Multiplex platforms are now designed to detect many rare diseases and mutations, but relying solely on these methods can affect the integrity and applicability of clinical genotyping assays. We used a novel approach to validate our genotyping platform by utilizing samples from the 1000 Genomes Project. As these samples are sequenced, the extent of information available allows us to validate many more genotype observations per sample, including both positive and negative calls. Leveraging the 1000 Genomes Project for validation expanded the number of biological samples available to use for validation of multiplex genotyping platforms. This not only addressed the limitations of current validation methods but also allowed for more accurate calculations of specificity. As a result, our panethnic, expanded carrier screening panel, inclusive of rare diseases and mutations, with demonstrated high sensitivity and specificity, can be confidently offered in a clinical setting.
Several professional societies have recognized the rationale for expanded, panethnic screening and have recently published a joint statement outlining criteria for the incorporation of such platforms into clinical practice for patients during pregnancy or before conception.5 These criteria include the following: (i) the included diseases should be severe enough to warrant consideration of prenatal diagnosis, (ii) data on carrier frequencies and detection rates is provided, and (iii) genetic counseling should be made available before and after testing. In spite of this guidance, however, there is much variability in expanded carrier screening across content, technology, and performance, making panel selection a challenging decision for clinicians. Currently available carrier screening panels are not standardized and include diverse diseases. Patients should be made aware of the types and clinical variability of diseases for which they are being screened. The available technologies for carrier screening, including chip-based genotyping and sequencing, all have inherent advantages and limitations. Ultimately, a combination of both technologies will provide the greatest reduction in residual risk. Regardless of platform, accurate validation of the platform is what will allow physicians to have confidence in results and thus drive their panel selection.
Although the combination of Coriell’s Subcollection of Heritable Diseases and the 1000 Genomes Project does allow for validation of a broader range of mutations, it is still not possible to validate all tested mutations because validation samples for certain mutations are not available through biorepositories. To account for this, we routinely use polymerase chain reaction and Sanger sequencing to confirm calls for variants that have not been validated previously as part of our expanded carrier screening process.
There are also assay-specific limitations to consider, such as polymorphisms near disease-causing mutations, triallelic variants, and pseudogenes. We have taken these limitations into consideration while developing our assay by designing multiple, bidirectional, sequence-specific probes for each mutation tested. This allows our assay to provide comprehensive coverage and accurate clinical results. While these design methods significantly increase the sensitivity and specificity of the assay, pseudogenes can still be particularly challenging because of their high sequence similarity. For instance, the false positive identified within the GBA gene in this study is likely a result of the presence of a GBA pseudogene (GBAP1). Therefore, as part of our expanded carrier screening process, mutations within genes with pseudogenes undergo a secondary bioinformatics review of the clustering algorithm to ensure accurate calls. This additional review is also applied for the analysis of triallelic variants and other unique cases.
In conclusion, we have introduced a novel validation method applicable to all multiplex platforms, using an easily accessible, highly accurate biorepository that allows for the validation of not only positive calls but also negative calls. On the basis of results from this study, we recommend including samples from the 1000 Genomes Project in the validation process of future multiplex platforms, which will increase confidence in their clinical use.
Disclosure
C.R., S.L.B., S.Y., S.R., N.K., and N.D.V. are employed by Recombine, Inc. C.R., S.L.B., S.Y., S.R., N.K., and N.D.V. have stock options in the company and hence are potential future shareholders in Recombine, Inc. A.B. and S.M. are shareholders in Recombine, Inc. B.C. and R.P. are employed by Reprogenetics, LLC.
Change history
22 October 2015
The authors have requested a retraction of the above article based on an error in the analysis of sample number.
References
Guttmacher AE, Collins FS. Genomic medicine—a primer. N Engl J Med 2002;347:1512–1520.
Emery A, Rimoin DL. Principles and Practice of Medical Genetics. 5th edn. New York: Churchill Livingston, 2007.
Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF. Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc 2001;76:777–783.
Costa T, Scriver CR, Childs B. The effect of Mendelian disease on human health: a measurement. Am J Med Genet 1985;21:231–242.
Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol 2015;125:653–662.
Tayo BO, Teil M, Tong L, et al. Genetic background of patients from a university medical center in Manhattan: implications for personalized medicine. PLoS One 2011;6:e19166.
Srinivasan BS, Evans EA, Flannick J, et al. A universal carrier test for the long tail of Mendelian disease. Reprod Biomed Online 2010;21:537–551.
Umbarger MA, Kennedy CJ, Saunders P, et al. Next-generation carrier screening. Genet Med 2014;16:132–140.
Jarvis M, Iyer RK, Williams LO, et al. A novel method for creating artificial mutant samples for performance evaluation and quality control in clinical molecular genetics. J Mol Diagn 2005;7:247–251.
Christensen TM, Jama M, Ponek V, et al. Design, development, validation, and use of synthetic nucleic acid controls for diagnostic purposes and application to cystic fibrosis testing. J Mol Diagn 2007;9:315–319.
Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–1073.
Abecasis G. Completion of the 1000 genomes project: results, lessons learned and open questions. 64th Annual Meeting of the American Society of Human Genetics, San Diego, CA, USA, 18-22 October 2014.
American College of Obstetricians and Gynecologists. Newborn screening. Obstet Gynecol 2007;110:1497–1500.
Jewish Genetic Disease Consortium. http://www.jewishgeneticdiseases.org/. Accesed 5 September 2014.
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006.
Acknowledgements
The authors thank the entire Bioinformatics and Product Teams at Recombine for all their help and support, and all laboratory personnel for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(PDF 74 kb)
Supplementary Table S2
(PDF 64 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Rivera-Garcia, C., Bristow, S., Yarnall, S. et al. Retracted: Validation of a multiplex genotyping platform using a novel genomic database approach. Genet Med (2015). https://doi.org/10.1038/gim.2015.101
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/gim.2015.101
