
Abstract
Purpose:
The goals of our study were to develop a noninvasive prenatal test for autosomal recessive monogenic conditions and to prove its overall feasibility and potential for clinical integration.
Methods:
We recruited a pregnant woman and her spouse, who had a proband child suffering from congenital deafness, and obtained the target-region sequencing data from a semicustom array that used genomic and maternal plasma DNA from three generations of this family. A haplotype-assisted strategy was developed to detect whether the fetus inherited the pathogenic mutations in the causative gene, GJB2. The parental haplotype was constructed using a trio strategy through two different processes, namely, the grandparent-assisted haplotype phasing process and the proband-assisted haplotype phasing process. The fetal haplotype was deduced afterward based on both the maternal plasma sequencing data and the parental haplotype.
Results:
The accuracy levels of paternal and maternal haplotypes obtained by grandparent-assisted haplotype phasing were 99.01 and 97.36%, respectively, and the proband-assisted haplotype phasing process yielded slightly lower accuracies of 98.73 and 96.79%, respectively. Fetal inheritance of the pathogenic gene was deduced correctly in both processes.
Conclusion:
Our study indicates that the strategy of haplotype-based noninvasive prenatal testing for monogenic conditions has potential applications in clinical practice.
Genet Med 16 12, 972–976.
Similar content being viewed by others
Main
Since cell-free fetal DNA (cffDNA) was discovered in the maternal circulation, a variety of noninvasive prenatal tests have been developed to avoid the risk caused by traditional invasive sampling procedures.1,2,3,4,5,6 Sequencing-based noninvasive technologies focus on the cytogenetic level, are rapidly developing, and are clinically available.4,5 However, technologies for monogenic diseases are still at the experimental laboratory stage.7,8,9,10
In our previous study,10 we proposed a robust mathematical model that could accurately recover both fetal genotype and fetal haplotype in one step, achieving complete characterization of the fetal genome. However, that study was limited by the strict requirements for samples and was not confirmed by actual clinical samples.
In the current study, blood samples were collected from a pregnant woman and closely related members of three generations of her family, which included the grandparents, parents, and one proband sister, to carry out noninvasive prenatal testing for congenital deafness. We constructed the parental haplotypes using the trio strategy and successfully conducted prenatal testing for the fetal condition, assisted by the information obtained from either the grandparents or the proband. Our study provides a reliable practical method of noninvasive prenatal testing for congenital deafness using maternal plasma sequencing.
Materials and Methods
Sample collection and identification of causative mutations
We recruited a pregnant woman, her spouse, and three generations of her family, which included a proband daughter whose audiograms showed severe to profound bilateral hearing loss. Genetic counseling was given to the family, and prenatal testing was provided as an option. Considering the risks of congenital deafness of fetus, the parents decided to take our noninvasive prenatal test. All participants in this study were recruited and gave informed consent in accordance with the Declaration of Helsinki. Ethical approvals were granted by the respective institutional boards of all participating institutions. The peripheral blood and amniotic fluid were obtained from the pregnant woman at the 17th and 18th/19th weeks of gestation. Peripheral blood samples of four grandparents were collected for parental haplotype construction. Polymerase chain reaction and Sanger sequencing were applied to analyze the mutation type in the GJB2 gene (Supplementary Table S1 and Figures S1 and S2 online). The father and the paternal grandfather were c.299delAT carriers. The pregnant mother and the maternal grandfather were c.235delC carriers. The proband was a compound heterozygote for the mutations.
Library preparation and sequencing
Genomic DNA of both parents and all four grandparents extracted from peripheral blood was fragmented by the sonicator. Maternal plasma was isolated using a two-step centrifugation protocol. After completion of end repair and A-tailing processes, adapters were ligated to each end of the DNA fragments. A DNA bar code was introduced to each sample during the polymerase chain reaction for massively parallel sequencing. Target-region capture was performed by a custom-designed 181.37M NimbleGen EZ array (containing the whole exome, 1M tag single-nucleotide polymorphisms (SNPs), and the major histocompatibility complex region) according to the manufacturer’s instructions. Postcapture libraries were sequenced using the Illumina Hiseq 2000 platform with 90-bp paired-end sequencing.
Alignment and SNP calling
The paired-end sequencing reads were mapped to the human reference genome (Hg19, GRCh37) using SOAP2.11 The reads that could be mapped to multiple locations and that had been amplified during the polymerase chain reaction process were removed. Then we performed SNP calling using the SOAPsnp software in the target region.12 The filter criteria (coverage >8 and quality value >20) were set to guarantee the accuracy of the genomic genotype.
Estimation of circulating cffDNA concentration
At the loci that were homozygous in both parents, but had different genotypes, the fetal genotype was an obligate heterozygote based on Mendel’s laws. Thus, the fractional fetal DNA concentration in the maternal plasma could be calculated with the loci for which the mother’s genotype was homozygous and for which the fetal genotype was predicted to be heterozygous. It can be described as the ratio of two times the fetus-specific allele count obtained from the father to the total base count of the other alleles.7,10
Parental haplotype construction
We constructed the parental haplotype using a strategy of trios based on Mendel’s law. For the grandparent-assisted haplotype phasing (GAHP) process, the paternal haplotype was constructed with each trio of the parent and the grandparents. For the proband-assisted haplotype phasing (PAHP) process, the parental haplotypes were constructed with the trio of father, mother, and proband.
Inference of the fetal haplotype
We used the linkage relationship obtained from parental haplotypes and the base distribution calculated using plasma sequencing to deduce the inherited fetal haplotype. The probabilities of the candidate haplotype combinations were calculated for each locus. According to the recombination characteristics of the gametogenesis process, we calculated the transmission probabilities using the distance between the neighboring sites to establish a hidden Markov model.10 In the decoding process of this model, we used the Viterbi algorithm to find the most likely sequence of hidden states and to deduce the inherited haplotype and recombination events in the fetus.13
Results
Alignment and parental haplotype construction
After target-region capture, the ~20-fold to ~30-fold enrichment products were subjected to 90-bp paired-end sequencing (Supplementary Table S2 online). All reads were aligned to the human reference genome, and SNP calling was performed. According to the SNP information, the fraction of cffDNA concentration within the maternal plasma was estimated at 15.10%.
The parental haplotypes were constructed using two strategies: the GAHP process and the PAHP process. Using the GAHP, a 104.91-Mb region of parental haplotypes was successfully phased, with 149,628 candidate markers distributed throughout the whole genome. Using the PAHP, a 104.93-Mb region containing 168,167 candidate markers was phased successfully.
Deduction of the fetal haplotype inheritance
In the maternal plasma, we established the hidden Markov model chain using the parental haplotype and the plasma sequencing data ( Figure 1 ), in which the hidden states were the actual fetal genotypes and the observed states were mixtures of the sequencing depths of the maternal and fetal genotypes in the plasma.10 After a decoding process, we obtained the inherited haplotype and the recombination events in the fetus. Using the parental haplotype obtained from the GAHP, we successfully deduced 126,600 loci of the fetus and discovered that the fetal inherited haplotype had 33 times recombination, and the inherited maternal one had 139 times. Using the PAHP strategy, we recovered 146,103 loci, with 60 and 181 times recombination in inherited paternal and maternal haplotype (Supplementary Figure S4 and Table S3 online).

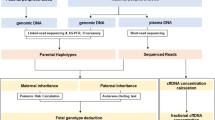
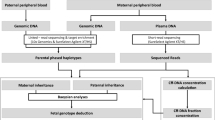
Flowchart of noninvasive prenatal testing strategy. (a) The genetic map of this recruited family. (b) Flowchart of the experiment and bioinformatics pipeline. cffDNA, cell-free fetal DNA; HMM, hidden Markov model.
To estimate the overall accuracy of the predicted fetal haplotype, we constructed the standard haplotype using the trio strategy with the information related to the parents and the cells of the amniotic fluid (Supplementary Table S3 online). Using the GAHP, 98.15% of the heterozygous loci of the paternal haplotype were inferred correctly in 67,978 loci, and 95.19% of the heterozygous loci of the maternal haplotype were inferred correctly in 69,346 loci. For the PAHP, the result was correct for 97.56% of the heterozygous loci of the paternal haplotype and 94.00% of the heterozygous loci of the maternal haplotype, about 1% lower than the results obtained with the GAHP strategy (Supplementary Tables S1 and S4 online).
Assessment of haplotype inference errors
The parental haplotype obtained from the PAHP process contained the recombination in the proband. Thus, the fetal haplotype deduced through PAHP had recombined twice. It showed higher recombination events as compared with the fetal haplotype deduced through GAHP, which may have generated more haplotype errors around recombination events. To further explore this influence of recombination events, we calculated the error rate of single-nucleotide variations in every 1-Mb region in the genome (Supplementary Figure S4 and Table S5 online). In the GAHP process, 23.87% of the errors in the paternal haplotype and 11.74% of the errors in the maternal haplotype were located within a 1-Mb distance around the break points. However, in the PAHP process, 30.12% of the errors in the paternal haplotype and 23.66% of the errors in the maternal haplotype were concentrated around the recombination break points of the fetus, and 9.83% of the errors in the paternal haplotype and 11.83% of the errors in the maternal haplotype were close to the proband recombination break points. Therefore, we concluded that the PAHP strategy enriched the recombination errors from the proband, which indicated a lower accuracy as compared with the GAHP strategy.
In addition to the influence of recombination events, the inference errors were also related to the parental haplotype phasing errors and the abnormal cffDNA concentration. In the parental haplotype phasing process, the inference errors may be caused by the low quality of SNP information of grandparents, parents, and proband, and would be superimposed in the phasing process. Thus, we used the haplotypes of the parents, proband, and fetus to reproduce the recombination process and found a few sporadic recombination points that only showed an isolated recombination signal in regions of less than 100kb. In the GAHP and PAHP strategies, about 29% of errors were related to these loci, which indicates that accurate determination of SNPs in genomic DNA is important to the whole analysis process. Unlike genomic DNA, the cffDNA in the maternal plasma was fragmented by natural degradation, characterized by an unstable portion of the human genome. We located the extreme cffDNA concentration loci (outside the 99% confidence interval of the genome-wide profile) and found that about 30% of the errors were close to them.
Noninvasive prenatal testing of congenital deafness
We conducted prenatal testing for congenital deafness based on the inherited haplotype in the GJB2 gene. Here, we defined the parental haplotypes as hap0 and hap1 to distinguish the pathogenicity. In GAHP, the paternal pathogenic allele inherited from the paternal grandfather was called f0, and the nonpathogenic allele was called f1. The maternal pathogenic allele was called m0, and the other was called m1. To encode the hidden Markov model, the fetal haplotype inheritance with regard to the GJB2 gene was f1 and m0, which indicated that the fetus was a heterozygous carrier of the c.235 delC mutation. In PAHP, because the proband was a typical patient with congenital deafness, we defined the parental alleles inherited by the proband as hap0, and the others were defined as hap1. Finally, the fetal haplotype in the GJB2 gene was also f1 and m0, which was consistent with the result from the GAHP strategy ( Figure 2 ). The diagnosis report of Chinese PLA General Hospital (Beijing, China) has supported our conclusion (Supplementary Figure S3 online). In this study, the genetic test was based on the analysis of fetal haplotype, instead of the detailed genotype.

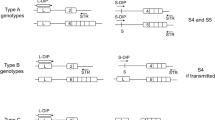
Results of noninvasive test for congenital deafness. (a) The inherited pathways of the parental alleles obtained by the GAHP strategy. (b) The inherited pathways of the parental alleles obtained by the PAHP strategy. (c) An enlarged view of the loci around the GJB2 gene using both the GAHP and PAHP strategies; both strategies show that the fetus inherited the hap f1 and hap m0 alleles. In all the graphs, the blue elements represent the parental alleles, and the red elements represent the maternal alleles (light colors indicate inheritance from the grandmothers, and dark colors indicate inheritance from the grandfathers). The lines below zero (black lines) indicate that the fetus inherited the pathogenic allele, and the lines above zero indicate that the fetus inherited the benign allele. GAHP, grandparent-assisted haplotype phasing; PAHP, proband-assisted haplotype phasing.
Discussion
About 1% of adults are carriers of mutant alleles.14 Among newborns, mutant alleles collectively account for ~20% of infant mortality and ~10% of pediatric hospitalizations.15 Effective diagnosis methods, especially prenatal diagnosis, need to be developed in order to improve the quality of life of the entire society. Congenital deafness is a common clinical genetic disorder, with an incidence of 1–3% in newborns.16 Early prenatal testing of hearing loss can give families more options for preparation and also allows time for laying the foundation for genetic therapy in the near future.17,18,19
In this study, we used the trio strategy with either grandparents or proband, incorporating the GAHP and PAHP methods. Both methods could determine the correct parental haplotypes to provide the linkage relationship in order to deduce the inherited haplotype of the fetus, although the accuracy of PAHP was lower because of the accumulation of recombination in the offspring. Thus, our method is appropriate for noninvasive prenatal testing of families with Mendelian diseases that can be deduced from the parental haplotypes of the grandparents or the proband. On the other hand, genomic DNA target sequencing requires no complicated experimental procedure, such as the previously reported haplotype-assisted methods, and is cost effective if the appropriate array is designed. Moreover, the turnaround time, including sampling process and testing on the HiSeq2500 platform, can be as short as 1 week, and the bioinformatics analysis can be accomplished within 1 day, which lends this type of procedure to large-scale clinical applications.8,9
However, there were still several imperfections of this method that should be considered. First, we constructed the parental haplotype based on the sequencing data of a trio in a single family, which greatly restricts the feasibility of our method for incomplete families for which these data are unavailable (although in most clinical cases, having an affected child is the reason for prenatal testing). Second, our testing was based on the definitive relevance between the disease and the related gene, so it can be used only for diseases for which the disease’s pathogenic gene has been fully explored. This is why our method is mainly applicable to Mendelian genetic disorders. Effective algorithms for the identification of de novo mutations must still be developed. There are also substantial ethical issues involved in noninvasive prenatal genome determination, especially as we progress to more comprehensive and convenient methods. However, there are numerous clinical scenarios in which this approach would be useful, including testing for fatal diseases or diseases that may lead to further medical complications.
Overall, we have proposed a promising noninvasive prenatal testing strategy for congenital deafness through massively parallel sequencing of maternal plasma. The haplotype-based approach described in this study may be extended to the noninvasive prenatal testing of most monogenic diseases.
Disclosure
X.L., H.G., F.C., Y.Z., W.X., X.P., S.C., P.L., C.Z., J.C., H.J., X.X., and W.W. are employees of BGI-Shenzhen. The other authors declare no conflict of interest.
References
Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997;350:485–487.
Lo YM, Tein MS, Lau TK, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 1998;62:768–775.
Chen S, Lau TK, Zhang C, et al. A method for noninvasive detection of fetal large deletions/duplications by low coverage massively parallel sequencing. Prenat Diagn 2013;33:584–590.
Dan S, Wang W, Ren J, et al. Clinical application of massively parallel sequencing-based prenatal noninvasive fetal trisomy test for trisomies 21 and 18 in 11,105 pregnancies with mixed risk factors. Prenat Diagn 2012;32:1225–1232.
Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR . Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA 2008;105:16266–16271.
Peters D, Chu T, Yatsenko SA, et al. Noninvasive prenatal diagnosis of a fetal microdeletion syndrome. N Engl J Med 2011;365:1847–1848.
Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91.
Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR . Non-invasive prenatal measurement of the fetal genome. Nature 2012;487:320–324.
Kitzman JO, Snyder MW, Ventura M, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012;4:137ra76.
Chen SP, Ge HJ, Wang XB, et al. Haplotype-assisted accurate noninvasive fetal whole genome recovery through maternal plasma sequencing. Genome Med 2013;5:18–27.
Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009;25:1966–1967.
Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 2009;19:1124–1132.
Tanaka H, Ishikawa M, Asai K, Konagaya A . Hidden Markov models and iterative aligners: study of their equivalence and possibilities. Proc Int Conf Intell Syst Mol Biol 1993;1:395–401.
Dürr A, Feingold J . [Genetic counseling for adults: the risk of late-onset inherited diseases]. Rev Prat 2011;61:535–538.
Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 2011;3:65ra4.
Hutchin T, Coy NN, Conlon H, et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK—implications for genetic testing. Clin Genet 2005;68:506–512.
Han B, Dai P, Qi QW, et al. [Prenatal diagnosis for hereditary deaf families assisted by genetic testing]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2007;42:660–663.
Nahar R, Puri RD, Saxena R, Verma IC . Do parental perceptions and motivations towards genetic testing and prenatal diagnosis for deafness vary in different cultures? Am J Med Genet A 2013;161A:76–81.
Chen W, Jongkamonwiwat N, Abbas L, et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 2012;490:278–282.
Acknowledgements
The study was supported by Shenzhen Birth Defect Screening Project Laboratory (JZF [2011] 861), Shenzhen Engineering Laboratory for Clinical Molecular Diagnostic (JZF [2011] 862), the National Natural Science Foundation of China (30972823), the Shanghai Health Bureau (XBR2011064), and the National Key Technology R&D Program (2006BAI05A09).
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
(DOC 2857 kb)
Rights and permissions
About this article

Cite this article
Meng, M., Li, X., Ge, H. et al. Noninvasive prenatal testing for autosomal recessive conditions by maternal plasma sequencing in a case of congenital deafness. Genet Med 16, 972–976 (2014). https://doi.org/10.1038/gim.2014.51
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.51
Keywords
This article is cited by
-
One-step noninvasive prenatal testing (NIPT) for autosomal recessive homozygous point mutations using digital PCR
Scientific Reports (2018)
-
Haplotype-based Noninvasive Prenatal Diagnosis of Hyperphenylalaninemia through Targeted Sequencing of Maternal Plasma
Scientific Reports (2018)
-
Non-invasive prenatal diagnosis of beta-thalassemia by semiconductor sequencing: a feasibility study in the sardinian population
European Journal of Human Genetics (2017)
-
Noninvasive prenatal diagnosis of 21-Hydroxylase deficiency using target capture sequencing of maternal plasma DNA
Scientific Reports (2017)
-
Development of novel noninvasive prenatal testing protocol for whole autosomal recessive disease using picodroplet digital PCR
Scientific Reports (2016)
