
Abstract
Purpose:
Autosomal recessive retinal dystrophies are clinically and genetically heterogeneous, which hampers molecular diagnosis. We evaluated identity-by-descent–guided Sanger sequencing or whole-exome sequencing in 26 families with nonsyndromic (19) or syndromic (7) autosomal recessive retinal dystrophies to identify disease-causing mutations.
Methods:
Patients underwent genome-wide identity-by-descent mapping followed by Sanger sequencing (16) or whole-exome sequencing (10). Whole-exome sequencing data were filtered against identity-by-descent regions and known retinal dystrophy genes. The medical history was reviewed in mutation-positive families.
Results:
We identified mutations in 14 known retinal dystrophy genes in 20/26 (77%) families: ABCA4, CERKL, CLN3, CNNM4, C2orf71, IQCB1, LRAT, MERTK, NMNAT1, PCDH15, PDE6B, RDH12, RPGRIP1, and USH2A. Whole-exome sequencing in single individuals revealed mutations in either the largest or smaller identity-by-descent regions, and a compound heterozygous genotype in NMNAT1. Moreover, a novel deletion was found in PCDH15. In addition, we identified mutations in CLN3, CNNM4, and IQCB1 in patients initially diagnosed with nonsyndromic retinal dystrophies.
Conclusion:
Our study emphasized that identity-by-descent–guided mutation analysis and/or whole-exome sequencing are powerful tools for the molecular diagnosis of retinal dystrophy. Our approach uncovered unusual molecular findings and unmasked syndromic retinal dystrophies, guiding future medical management. Finally, elucidating ABCA4, LRAT, and MERTK mutations offers potential gene-specific therapeutic perspectives.
Genet Med 16 9, 671–680.
Similar content being viewed by others


Introduction
Retinal dystrophies (RDs) have a profound impact on an individual’s life. Patients develop significant visual impairment, often leading to irreversible blindness, with an age of onset ranging from birth to late adulthood. RDs are inherited, with autosomal recessive (AR), autosomal dominant, and X-linked inheritance patterns described.1 RDs generally display a tremendous genetic heterogeneity: more than 240 disease loci (>200 genes) have been identified so far, each accounting for only a fraction of patients. Moreover, up to 50% of disease genes are still unidentified.2 In addition, retinal phenotypes often overlap and might be associated with systemic features developing in a later disease stage.3 Overall, this profound genetic and phenotypic complexity severely hampers the establishment of a molecular diagnosis, which is essential in view of not only genetic counseling but also gene therapy.4
Molecular diagnostics for RDs recently shifted from commercial microarray-based testing for known mutations and Sanger sequencing of limited gene sets5,6 to massively parallel sequencing (MPS), allowing fast and cost-effective sequencing of comprehensive RD gene panels.7,8,9 One step further is the implementation of whole-exome sequencing (WES), in which all (coding) exons are enriched and sequenced, enabling the identification of mutations in known as well as novel RD genes. However, the enormous number of variants thus identified is extremely challenging to interpret.
Here, homozygosity or identity-by-descent (IBD) mapping offers an excellent filtering strategy because this technique pinpoints the location of the causal gene in inbred families in which an AR disease is segregating. IBD mapping is based on the concept that consanguineous individuals have a high risk of carrying two identical disease alleles originating from a common ancestor. The region encompassing the disease gene is homozygous and shared between different affected individuals of the same family.10 IBD mapping has played a major role in both RD gene discovery and routine screening, combined with both Sanger sequencing and MPS.5,11,12
In this study, we performed IBD mapping and subsequent Sanger sequencing or WES in 26 consanguineous families with a nonsyndromic (19) or syndromic (7) AR RD. In addition to a high detection rate, our approach identified conventional subtle mutations as well as a large deletion. In addition, WES was instrumental in the identification of mutations located both within and outside IBD regions. Moreover, the molecular diagnosis revealed a complex clinical diagnosis in three families, directly influencing clinical follow-up and management. Finally, elucidating ABCA4, LRAT, and MERTK mutations offers gene-specific therapeutic perspectives.
Materials and Methods
Subject enrollment and clinical evaluation
Thirty-seven individuals from 26 consenting families were recruited from the Center for Medical Genetics Ghent and the Department of Ophthalmology, Ghent University Hospital, Ghent, Belgium; the Department of Ophthalmology, Queen Fabiola Children’s University Hospital, Brussels, Belgium; and the Department of Ophthalmology and Center for Human Genetics, Leuven University Hospitals, Leuven, Belgium. Informed consent was obtained and the research protocols adhered to the tenets of the Declaration of Helsinki. Patients had histories and pedigree analysis taken and underwent ophthalmic examination, including best-corrected visual acuity testing, slit-lamp examination, Goldmann visual field testing, dilated fundus examination, and full-field flash electroretinography.13 Nineteen probands were initially diagnosed with one of the following nonsyndromic RDs: Leber congenital amaurosis (LCA) (4), early-onset RD (EORD) (3), AR retinitis pigmentosa (ARRP) (9), or AR cone-rod dystrophy (ARCRD) (3). In addition, seven probands were referred for a complex RD: Senior–Loken syndrome (2), LCA in combination with intellectual disability (ID) (2) or deafness (1), and Usher syndrome type I (1) or II (1).
Genetic prescreening, Sanger sequencing, and WES were performed on each proband (first child affected in the family). Following the identification of a causal mutation, medical records were reassessed.
Genetic prescreening
Genomic DNA was extracted from leukocytes (Puregene DNA isolation kit, Gentra, Qiagen, Venlo, The Netherlands). Depending on the RD, probands underwent the following prescreening prior to this study: LCA: LCA Arrayed Primer EXtension (APEX) chip (Asper Ophthalmics, Tartu, Estonia), followed by Sanger sequencing of CEP290, CRB1, RPE65, AIPL1, GUCY2D, and CRX;14 ARRP: ARRP APEX chip; and ARCRD: ARRP or ABCA4 APEX chip. In addition, the probands of F1, F13, and F15 were included in MPS of 16 LCA genes.7 Table 1 contains information on patient-specific prior genetic screening.
IBD mapping
Genome-wide IBD mapping was performed in 37 individuals using Affymetrix GeneChip Human Mapping 250K Nsp arrays (DNAVision, Charleroi, Belgium). Single-nucleotide polymorphism (SNP) genotypes were analyzed with an in-house Perl script and with PLINK.15 Resulting homozygous regions were evaluated for their size and number of SNPs. Regions were excluded if unaffected siblings were homozygous for the same genotype as the affected individuals. Of note, this may result in incorrect exclusion of regions in the case that the affected sibling shows an overlapping identity-by-state interval.
Sequencing of candidate genes and WES
At the onset of this study, 16 families underwent IBD-guided Sanger sequencing. Genes were prioritized based on their location in homozygous regions and association with AR RDs.2 Primers were designed using Primer3Plus16 to cover all coding exons and intron–exon boundaries. Sanger sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Life Technologies Europe, Ghent, Belgium) on a 3730xl DNA Analyzer (Applied Biosystems).
Following the introduction of MPS technology, IBD-guided WES was performed in the probands of 10 additional families. Exome enrichment and sequencing were performed by Aros AB (Aarhus, Denmark) using the Illumina TruSeq Exome Enrichment Kit (Illumina, San Diego, CA) (F13-F16, F18, F20, F21) and the Nextera Rapid Capture Expanded Exome kit (F12, F17, F19), followed by paired-end sequencing on a HiSeq 2000 (2 × 100 cycles). For the TruSeq kit, six indexed samples were pooled and sequenced across two flow cell lanes. For the Nextera kit, 12 indexed samples were pooled and sequenced across four flow cell lanes.
The CLC Genomics Workbench v6.0.4 (CLC bio, Cambridge, MA) was employed for read mapping against the human genome reference sequence (NCBI, GRCh37), postmapping duplicate read removal, coverage analysis, and quality-based variant calling. All mutations were confirmed with Sanger sequencing. Novel sequence variants were evaluated using the Alamut mutation interpretation software v2.2.1 (Interactive Biosoftware, Rouen, France), Alamut HT v1.1.5 software, literature search, and segregation analysis in all available individuals.
Mutation nomenclature uses numbering with the A of the initiation codon ATG as +1 (http://www.hgvs.org/mutnomen), based on the following RefSeqs: NM_000350.2 (ABCA4), NM_001030311.2 (CERKL), NM_001042432.1 (CLN3), NM_020184.3 (CNNM4), NM_201253.2 (CRB1), NM_001029883.1 (C2orf71), NM_001142800.1 (EYS), NM_001023570.2 (IQCB1), NM_004744.3 (LRAT), NM_006343.2 (MERTK), NM_022787.3 (NMNAT1), NM_000283.3 (PDE6B), NM_152443.2 (RDH12), NM_020366.3 (RPGRIP1), and NM_206933.2 (USH2A).
Copy-number variation screening and deletion delineation
Long-range amplicons were amplified with the iProof High-Fidelity DNA Polymerase (Bio-Rad, Nazareth Eke, Belgium) and evaluated on the LabChip GX (Caliper, PerkinElmer, Zaventem, Belgium; HT DNA 5K kit).
Multiplex ligation-dependent probe amplification was performed using the SALSA MLPA P328 EYS probemix (MRC Holland, Amsterdam, The Netherlands) following the manufacturer’s protocols.
Results
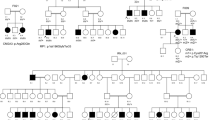
The PLINK software was used for the detection of homozygous regions equal to or above 1 Mb in size. Following IBD mapping, known RD genes2 were prioritized for Sanger sequencing in 16 families, whereas 10 families underwent WES ( Figure 1 ). This revealed causal mutations in known RD genes in 20/26 families ( Table 1 ). Supplementary Table S1 online contains in silico predictions for all missense mutations, whereas Supplementary Figure S1 online shows segregation analysis. Figures 2 and 3 contain a selection of interesting molecular and clinical findings, respectively. In the following sections, results are discussed based on the applied approach (IBD + Sanger sequencing or IBD + WES).

Flowchart of the study. Unusual clinical findings and molecular causes are indicated in bold. ARCRD, autosomal recessive cone-rod dystrophy; ARRP, autosomal recessive retinitis pigmentosa; EORD, early-onset retinal dystrophy; IBD, identity-by-descent; ID, intellectual disability; LCA, Leber congenital amaurosis; RD, retinal dystrophy; SLS, Senior–Loken syndrome; USH, Usher syndrome.

Unusual molecular and clinical findings revealed in this study. (a) Sanger sequencing of the CNNM4 gene, located in the largest IBD region of F7, revealed a novel frameshift mutation. Based on these molecular data, dental clinical records were revisited, showing features typical for dentinogenesis imperfecta: yellowish discoloration and quality loss of enamel, especially at tips of teeth, as well as fang-like malformation of canines (VI:10, age 3 years 10 months). Fundus picture of the left eye of patient VI:2 age 15 years 10 months: note yellowish discoloration of macula, and outer retinal atrophy with intraretinal pigmentation in (mid)periphery; macula around area of central atrophy has better preserved retinal quality. (b) Delineation of the homozygous PCDH15 deletion in the proband of F11. Amplicons for which PCR amplification failed are indicated in green. The deletion, containing exons 1 and 2 of the PCDH15 gene, was refined by two subsequent rounds of PCR walking, resulting in an estimated deleted region of 163.5 to 165.7 kb. This region was amplified by long-range PCR (purple bar) and sequenced, delineating the 164 kb deletion at the nucleotide level (green bar). (c) Combined IBD mapping and WES identified a novel nonsense mutation in the CLN3 gene in the proband of F14 (IV:4), initially diagnosed with EORD. Revisiting of the clinical record revealed psychiatric symptoms compatible with Batten disease. Predictive testing of the youngest sister (IV:6) uncovered her disease status. Individuals who underwent IBD mapping are indicated with an asterisk. ARCRD, autosomal recessive cone-rod dystrophy; EORD, early-onset retinal dystrophy; IBD, identity-by-descent; PCR, polymerase chain reaction; USH, Usher syndrome; WES, whole-exome sequencing.

Clinical phenotype of five retinol dystrophy patients with an established molecular diagnosis. Proband of F2, VI:2 (LRAT), left eye: fundus picture of left eye of proband of F2 with EORD due to biallelic mutations in LRAT. Typical grayish patches of outer retinal atrophy in peripheral macula and retinal periphery with much better preserved macula with some degree of preretinal fibrosis but otherwise without intraretinal pigment migration, at age 17 years. Picture typical for phenotype of LRAT-related EORD. Proband of F9, II:1 (IQCB1), right eye: fundus picture of right eye of proband of F9 with SLS due to biallelic mutations in IQCB1. Peripheral grayish patches in retinal periphery typical of outer retinal atrophy, with some minor intraretinal pigmentation, with relatively slightly better preserved macula, at age 17 years. Proband of F10, II:1 (IQCB1), right eye: fundus picture of right eye of proband of F10 with SLS due to biallelic mutations in IQCB1. Peripheral grayish patches of outer retinal atrophy, with spicular intraretinal pigmentation and narrowing of the retinal vasculature, with relatively much better preserved macula, at age 20 years. Proband of F10, II:1 (IQCB1), right eye: blue-light autofluorescence image of proband of F10 showing mild hyperautofluorescent central perimacular ring. Two affected brothers of F20, IV:3 and IV:6 (CERKL), right macula: intrafamilial phenotypical variability can be seen in pictures of right macula of proband of F20 (IV:3), with ARCRD due to biallelic CERKL mutations, showing relatively mild atrophy in the central macula, whereas the right macula of his older brother (IV:6) is completely atrophic, but for one minute central foveal island. ARCRD, autosomal recessive cone-rod dystrophy; EORD, early-onset retinal dystrophy; SLS, Senior–Loken syndrome.
Combined IBD mapping and Sanger sequencing
LCA and EORD. Using IBD-guided Sanger sequencing, one LCA and one EORD family were resolved out of three families (1 EORD, 2 LCA) ( Figure 1 , Table 1 ). In the first family (F1, LCA), a novel homozygous nonsense mutation was identified in RDH12: c.912G>A (p.Trp304*).7 This mutation is located in the last exon and thus not expected to induce nonsense-mediated decay. The proband was diagnosed with LCA at the age of 2 years and 3 months based on severe visual deficiency from birth, congenital nystagmus, hyperopia, and night blindness. An electroretinography performed at the age of 2 years and 7 months showed absent rod-specific and only residual cone-specific responses. Subsequent segregation analysis demonstrated that the apparently unaffected, younger brother of the proband (IV:3) is also homozygous (see Supplementary Figure S1 online) and will develop a similar phenotype. In the second family (F2, EORD), the result of Sanger sequencing of IMPG2, which is located in the largest IBD region, was negative, whereas Sanger sequencing of LRAT, which is located in the region with the second-largest number of consecutive SNPs, identified homozygosity for a novel missense mutation, c.326G>T (p.Arg109Leu) (see Supplementary Table S1 online) ( Figure 3 ).
Autosomal recessive retinitis pigmentosa. The molecular defect was identified in 4/5 ARRP families using combined IBD mapping and Sanger sequencing ( Figure 1 ). The causal mutation was located in the largest (F3, F5, F6) or second-largest (F4) IBD region ( Table 1 ).
In families F3, F4, and F5, mutations in RDH12 are responsible for the phenotype. The causal mutation in F3 and F4 is a known missense mutation (c.464C>T, p.Thr155Ile; see Supplementary Table S1 online), shown to reduce the conversion of all-trans retinal to all-trans retinol.17 No family members were available for segregation testing in F4. In F5, a novel missense mutation was identified, c.803G>T (p.Cys268Phe), which is predicted to affect protein function by a selection of prediction programs (Supplementary Table S1 online).
Finally, a homozygous missense mutation in USH2A (c.12574C>T, p.Arg4192Cys; see Supplementary Table S1 online) was detected in the proband of F6 following candidate gene screening of RD3 and USH2A—both located in the largest IBD region. A different missense variant (c.12575G>A, p.Arg4192His) has previously been described at the same amino acid position.18 However, the latter variant is predicted to be benign (data not shown).
Autosomal recessive cone-rod dystrophy. This study included three families with ARCRD ( Figure 1 ). Combined IBD mapping and Sanger sequencing revealed disease-causing mutations in two families (F7 and F8).
In F7, sequencing of MERTK and CNNM4, located in the two largest IBD regions, identified a novel homozygous frameshift mutation in CNNM4, c.189del (p.Asp63Glufs*12). Because CNNM4 mutations are associated with Jalili syndrome (OMIM 217080), characterized by CRD and amelogenesis imperfecta,19,20 dental records were revisited, and these confirmed amelogenesis imperfecta in all affected family members ( Figure 2a ).
The second family (F8) contained one sporadic CRD patient, who tested negative for the ABCA4 chip ( Table 1 ). However, the ABCA4 gene was located in the region with the largest number of consecutive homozygous SNPs. Subsequent ABCA4 Sanger sequencing revealed a novel homozygous frameshift mutation (c.3819dup, p.Leu1274Serfs*8).
Syndromic RDs. In total, patients from seven families diagnosed with a syndromic RD were enrolled. Six of them were subjected to Sanger sequencing following IBD mapping ( Figure 1 ).
In two cases, known null mutations in IQCB1 were found to underlie Senior–Loken syndrome (F9: c.1381C>T (p.Arg461*)21; F10: c.424_425del (p.Phe142Profs*5); ( Figure 3 ).21 Interestingly, in F9, only the youngest (II:2) of the affected brothers had been diagnosed with Senior–Loken syndrome and ID prior to his recent death, suggesting either the involvement of modifying factors or an independent AR entity.
The proband of F11 suffered from RP and congenital, bilateral, and profound sensorineural hearing loss, and did not develop speech. The fifth-largest IBD region contained the PCDH15 gene, which is known to be associated with Usher syndrome type 1 (USH1F, OMIM 602083). Sanger sequencing of all exons revealed an inability to amplify the first two exons of PCDH15, indicative of a homozygous deletion. Figure 2b shows the delineation of this deletion. First, polymerase chain reaction walking with 13 amplicons delineated the deletion to a region ranging from 139.2 to 167.0 kb. A second round of refinement using 20 additional amplicons reduced this interval to 163.5–165.7 kb, allowing long-range polymerase chain reaction and Sanger sequencing of the junction product. This revealed a homozygous deletion of 164 kb with a 2-bp region of microhomology (AT) at the junction (chr10:g.56408521-g.56572561). Of note, the IBD mapping data contained a region of no calls overlapping with this region.
Combined IBD mapping and WES
Following IBD mapping, WES was performed in the probands of 10 families: 2 families with LCA (F12, F13), 2 families with EORD (F14, F15), 4 families with ARRP (F16–F19), 1 family with ARCRD (F20), and 1 family with LCA and ID (F21) ( Figure 1 ). For each sample, at least 73% of unique reads were aligned against the human genome. An average coverage of 60–98× was achieved for the regions enriched by both exome kits. Between 87 and 94% of these regions had a read depth of at least 10× (see Supplementary Table S2 online).
Variants were called heterozygous or homozygous in case of a variant allele frequency of 20–70% and ≥70%, respectively. Variant filtering was performed in two steps. First, homozygous variants located in major IBD regions or known RD genes were selected (see Supplementary Table S3 online). Subsequently, variants were selected based on their predicted translation effect and in silico predicted effect on protein function using Alamut HT (see Supplementary Table S4 online).
Variant filtering based on IBD regions. Variant filtering based on IBD regions revealed the causal genetic defect in five families. In the proband of F12, a sporadic patient with LCA, several small homozygous regions were identified, suggesting a distant common ancestor of both parents (see Supplementary Table S3 online). In the largest IBD region, a novel homozygous nonsense mutation was identified in the RPGRIP1 gene, c.2758C>T (p.Gln920*).
In F14 (EORD), a novel nonsense mutation was identified in the CLN3 gene (c.784A>T, p.Lys262*) ( Figure 2c ). Mutations in CLN3 are associated with Batten disease or juvenile neuronal ceroid lipofuscinosis (OMIM 204200). Patients most often present with progressive visual loss between the ages of 5 and 10, followed by loss of motor coordination, mental decline, and seizures, starting at the age of 10–12 and leading to premature death. The majority of CLN3 mutations produce prematurely truncated products and cause “classic” juvenile neuronal ceroid lipofuscinosis. However, some mutations cause a protracted course of juvenile neuronal ceroid lipofuscinosis.22 The proband of F14 was referred for genetic screening for EORD at the age of 10, when neurologic symptoms were not yet evident. Revisiting of the clinical records uncovered psychiatric manifestations from the age of 12 that are in keeping with a diagnosis of Batten disease. Segregation analysis showed that the youngest sister of the proband, aged 5, is also homozygous for the mutation, revealing a presymptomatic molecular diagnosis ( Figure 2c ).
In the third family, F16 (ARRP), filtering of the exome variants with the largest IBD region revealed only two novel or rare missense variants predicted to affect protein function, one of which was c.2180G>A (p.Arg727Gln) in the MERTK gene (see Supplementary Table S1 online). The MERTK Arg727 residue is known to be involved in a hydrogen bond with the hydroxyl group of the ligand compound 52 (ref. 23).
In the fourth family, F17 (ARRP), a homozygous frameshift mutation was identified in C2orf71, c.1764del (p.Glu589fs). Segregation analysis showed that both parents and the older sibling are heterozygous carriers of the C2orf71 mutation, whereas the younger sister is also homozygous (see Supplementary Figure S1 online).
In the fifth family, F18 (ARRP), previous Sanger sequencing of four RD genes (LRAT, RP1, ADAM9, CNGA1) located in the five largest IBD regions did not identify any causal mutations. Because of this, a broader selection of 13 IBD regions was made for exome filtering (see Supplementary Table S3 online). In the 11th IBD region, a homozygous indel mutation was identified in PDE6B, c.121_125delins15 (p.Pro41Glufs*128; see Supplementary Table S4 online). The CLC software initially reported this mutation as c.126_127insAG (p.Asp43fs). No family members were available for segregation analysis.
Variant filtering based on known RD genes. Analysis of all variants located in the largest IBD regions identified in F13, F15, and F19–F20 did not reveal obvious causal mutations. In addition, despite presumed consanguinity, no large IBD regions were identified in the proband of F21. In these five families, variant filtering was performed against a list of all RetNet genes associated with both isolated as well as syndromic RDs.2 Both homozygous and heterozygous variants were taken into account. Overall, this yielded on average 173 coding variants in the 209 genes per patient.
In the proband of F13 (LCA), 16 novel or rare missense changes were predicted to affect protein function by at least 2/4 in silico prediction programs (see Supplementary Table S4 online). One of these was a known heterozygous missense mutation in NMNAT1, c.205A>G (p.Met69Val).24 Subsequently, we evaluated NMNAT1 variants in more detail and identified a second heterozygous missense mutation, c.769G>A (p.Glu257Lys), which is a known mutation not predicted to affect protein function (see Supplementary Table S1 online).24
In F15 (EORD), we identified a heterozygous known frameshift mutation in EYS (c.9368dup, p.Asn3123Lysfs*3).25 Subsequent Sanger sequencing of exons with a coverage below 20× did not identify a second mutation. As several copy-number variations (CNV) have been described in EYS,26 multiplex ligation-dependent probe amplification was performed. However, no CNV was detected.
The proband of F19 (ARRP) harbors several large IBD regions, suggesting close parental consanguinity (see Supplementary Table S3 online). First, Sanger sequencing was performed for MERTK, CEP290, FAM161A, and SNRNP200, which are located in the two largest IBD regions. Because no mutations were found, the proband was subjected to WES. The application of the RetNet filter allowed the identification of the following homozygous missense mutation in the USH2A gene: c.10721G>T (p.Gly3574Val; see Supplementary Table S1 online). This mutation, together with the known splice-site mutation c.3317-2A>G, has been described in a sporadic ARRP patient.9 Interestingly, it is located in a homozygous region of only 1.5 Mb, in which the USH2A gene is only partially located (see Supplementary Figure S2 online).
In F20 (ARCRD), RetNet-based variant filtering revealed 170 variants resulting in an amino acid change. Of the six truncating variants, one was the known mutation c.847C>T (p.Arg283*)27 in the CERKL gene, which is located in the 38th IBD region ( Figure 3 ). Subsequent Sanger sequencing confirmed that the proband of F20 is homozygous for this CERKL mutation.
Variant filtering against the RetNet genes in the proband of F21 (LCA + ID) identified a novel homozygous frameshift mutation in IQCB1 (c.1241_1242del, p.Leu414Hisfs*4). IQCB1 mutations have been associated with Senior–Loken syndrome21 as well as LCA.28 When revisiting the patient’s medical records, renal involvement with nephronophthisis was observed.
In conclusion, a variant-filtering strategy based on either IBD regions or RetNet genes allowed identification of causal mutations in 9/10 exomes.
Discussion
The tremendous genetic and phenotypic heterogeneity of RDs poses a major challenge for establishing a molecular diagnosis. Here, we evaluated an integrated approach based on IBD-guided Sanger sequencing or WES to identify the causal gene in 26 consanguineous RD families.
The combination of IBD mapping and Sanger sequencing proved to be effective, as this strategy resulted in the identification of the causal mutation in 11/16 families (69%) ( Figure 1 ). In 10/11 cases, the causal gene was located in the first- or second-largest homozygous region ( Table 1 ). It has previously been observed that the causal gene resides in the largest homozygous region in about 17% of consanguineous families from inbred populations.29 In the remaining group, however, gene-by-gene Sanger sequencing tends to be laborious and expensive.
In these patients, WES represents a more efficient way toward a molecular diagnosis. Indeed, using WES, we were able to identify causal genes located not only in high-ranked but also in markedly lower-ranked regions in four families (F18–F21, Table 1 ). Moreover, WES allowed the identification of a compound heterozygous genotype (F13), one of the pitfalls of IBD mapping. In total, WES revealed mutations in 9/10 families ( Figure 1 ).
This study showed that combined IBD mapping and WES can lead to successful albeit challenging mutation discovery in a single individual of consanguineous origin, which is in agreement with a similar large-scale study in RD combining WES with IBD mapping12 and two studies applying WES in RD.30,31 Given the large number of coding variants identified through WES, comprehensive variant filtering is of utmost importance. Variant filtering based on IBD regions or RetNet genes drastically reduced variant numbers (see Supplementary Tables S4 and S5 online). Interestingly, our data demonstrated that RetNet gene–based filtering would have been superior to IBD filtering as a first-pass filter for several reasons. First, the RetNet filter generated markedly fewer variants to interpret, with an average of 173 heterozygous or homozygous coding variants per proband for the 209 RD genes assessed here—in contrast to up to 548 homozygous coding variants located in the IBD regions. Second, the RetNet filter allowed identification of a compound heterozygous genotype (F13) and mutations located in very small homozygous regions (F19–F21), which were not picked up by the IBD filter. Third, all mutations identified through the IBD filter (F12, F14, F16–F18) would have also been identified using the RetNet gene filter. These insights, together with efforts to apply IBD mapping directly to exome data,32 will eliminate the need for prior genome-wide SNP genotyping in future diagnostic WES applications in RD. However, for those families not having access to WES, IBD-guided targeted resequencing of a selected number of genes using a small MPS benchtop sequencer will still be cost effective in consanguineous families.
One of the key findings of this study is the identification of mutations in syndromic RD genes that would not have been selected for routine first-pass screening based on the patient’s initial diagnosis. Indeed, in syndromic RDs the retinal phenotype is often one of the first to arise, prior to other, later-onset, systemic manifestations. This is illustrated by the identification of a homozygous CNNM4 frameshift mutation in a family initially referred to us with ARCRD (F7, Figure 2a ). It was only after review of clinical dental records that the presence of amelogenesis imperfecta was noticed, in agreement with Jalili syndrome.19,20 Second, the identification of a homozygous CLN3 nonsense mutation in a proband initially diagnosed with EORD uncovered a link with emerging psychiatric and neurodegenerative manifestations with an important, unexpected impact on her clinical prognosis (F14, Figure 2c ). Third, the identification of a truncating IQCB1 mutation in a patient initially diagnosed with LCA and ID implies close renal follow-up. Of note, the detection of homozygous RDH12, CLN3, and C2orf71 mutations in unaffected siblings of F1, F14, and F17, respectively, had important implications for clinical follow-up.
In addition to unexpected clinical and prognostic findings, our IBD-based approach revealed unusual molecular findings such as a large genomic rearrangement in PCDH15 ( Figure 2b ). Besides a large IBD region encompassing this gene, the homozygous deletion appeared in the SNP data as a no call region. PCDH15 large rearrangements are a known cause of Usher syndrome type 1. So far, six deletions with nonrecurrent breakpoints have been described, all removing at least one exon at the 5' end of the gene.33,34,35 It is presumed that the occurrence of distinct deletions in this region is related to the large size of PCDH15, in particular of the intronic regions between the first exons. This is in line with our findings, as bioinformatic analysis of the breakpoint regions suggests nonhomologous end joining or fork stalling and template switching as the most likely mechanisms for this novel deletion (data not shown). Although only a limited number of CNVs have been described in RDs, not surprisingly with low frequencies comparable to other single-base RD mutations,26,36 their occurrence warrants implementation of a CNV analysis algorithm in addition to IBD data analysis.
Overall, our approach revealed mutations in 20/26 (77%) families in the following 14 known RD genes: ABCA4, CERKL, CLN3, CNNM4, C2orf71, IQCB1, LRAT, MERTK, NMNAT1, PCDH15, PDE6B, RDH12, RPGRIP1, and USH2A. This overall mutation detection rate is in agreement with reported contributions of RD genes. In addition, the numbers of unique mutations and causal genes identified here illustrate the allelic and locus heterogeneity, respectively.
So far, the causal gene cannot be identified in six families. We anticipate these will likely be among “gene discovery” families, as known RD genes account for only 70–80% of cases. The combination of IBD mapping, WES, and recent variant filter tools specifically designed for RDs37 provides a strong asset in gene identification in such families.12 Of note, we cannot rule out the presence of complex rearrangements and regulatory or deep-intronic mutations in known RD genes, taking into account the limitations of WES in detecting CNVs and enriching these regions, respectively. Whole-genome sequencing will offer a more suitable detection technique for these mutation mechanisms.38
In conclusion, our study emphasized that IBD-guided mutation analysis and/or WES is a powerful tool for the molecular diagnosis of RD—even in a single individual. We unmasked unusual molecular and consequent clinical findings—the latter guiding future medical management and prognosis. Finally, elucidating ABCA4, LRAT, and MERTK mutations in three families offers potential gene-specific therapeutic perspectives.
Disclosure
The authors declare no conflict of interest.
References
den Hollander AI, Black A, Bennett J, Cremers FP . Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest 2010;120:3042–3053.
RetNet, the Retinal Information Network. http://www.sph.uth.tmc.edu/RetNet. Accessed July 25th, 2013.
Courtney RJ, Pennesi ME . Inherited retinal degenerations with systemic manifestations. Int Ophthalmol Clin 2012;52:119–147.
Cideciyan AV . Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 2010;29:398–427.
den Hollander AI, Roepman R, Koenekoop RK, Cremers FP . Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 2008;27:391–419.
Stone EM . Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol 2007;144:791–811.
Coppieters F, De Wilde B, Lefever S, et al. Massively parallel sequencing for early molecular diagnosis in Leber congenital amaurosis. Genet Med 2012;14:576–585.
Neveling K, Collin RW, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012;33:963–972.
Glöckle N, Kohl S, Mohr J, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 2014;22:99–104.
Lander ES, Botstein D . Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987;236:1567–1570.
Littink KW, den Hollander AI, Cremers FP, Collin RW . The power of homozygosity mapping: discovery of new genetic defects in patients with retinal dystrophy. Adv Exp Med Biol 2012;723:345–351.
Abu-Safieh L, Alrashed M, Anazi S, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res 2013;23:236–247.
Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M ; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol 2009;118:69–77.
Coppieters F, Casteels I, Meire F, et al. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum Mutat 2010;31:E1709–66.
Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA . Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007;35(Web Server issue):W71–W74.
Thompson DA, Janecke AR, Lange J, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum Mol Genet 2005;14:3865–3875.
McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL . Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet 2010;47:499–506.
Parry DA, Mighell AJ, El-Sayed W, et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet 2009;84:266–273.
Polok B, Escher P, Ambresin A, et al. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am J Hum Genet 2009;84:259–265.
Otto EA, Loeys B, Khanna H, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 2005;37:282–288.
Kousi M, Lehesjoki AE, Mole SE . Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63.
Huang X, Finerty P Jr, Walker JR, et al. Structural insights into the inhibited states of the Mer receptor tyrosine kinase. J Struct Biol 2009;165:88–96.
Falk MJ, Zhang Q, Nakamaru-Ogiso E, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet 2012;44:1040–1045.
Audo I, Sahel JA, Mohand-Saïd S, et al. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat 2010;31:E1406–E1435.
Pieras JI, Barragán I, Borrego S, et al. Copy-number variations in EYS: a significant event in the appearance of arRP. Invest Ophthalmol Vis Sci 2011;52:5625–5631.
Tuson M, Marfany G, Gonzàlez-Duarte R . Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am J Hum Genet 2004;74:128–138.
Estrada-Cuzcano A, Koenekoop RK, Coppieters F, et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci 2011;52:834–839.
Woods CG, Cox J, Springell K, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet 2006;78:889–896.
Chen Y, Zhang Q, Shen T, et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Invest Ophthalmol Vis Sci 2013;54:4351–4357.
Corton M, Nishiguchi KM, Avila-Fernández A, et al. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS ONE 2013;8:e65574.
Rödelsperger C, Krawitz P, Bauer S, et al. Identity-by-descent filtering of exome sequence data for disease-gene identification in autosomal recessive disorders. Bioinformatics 2011;27:829–836.
Aller E, Jaijo T, García-García G, et al. Identification of large rearrangements of the PCDH15 gene by combined MLPA and a CGH: large duplications are responsible for Usher syndrome. Invest Ophthalmol Vis Sci 2010;51:5480–5485.
Le Guédard S, Faugère V, Malcolm S, Claustres M, Roux AF . Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol Vis 2007;13:102–107.
Roux AF, Faugère V, Le Guédard S, et al.; French Usher Syndrome Collaboration. Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J Med Genet 2006;43:763–768.
Wissinger B, Schaich S, Baumann B, et al. Large deletions of the KCNV2 gene are common in patients with cone dystrophy with supernormal rod response. Hum Mutat 2011;32:1398–1406.
Wagner AH, Taylor KR, DeLuca AP, et al. Prioritization of retinal disease genes: an integrative approach. Hum Mutat 2013;34:853–859.
Nishiguchi KM, Tearle RG, Liu YP, et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA 2013;110:16139–16144.
Acknowledgements
This work was supported by grants from the Research Foundation Flanders (FWO) and Bijzonder Onderzoeksfonds (BOF) (FWO/KAN/31509107; FWO11/KAN/013-31524611; FWO 3G079711; BOF10/STA/055 to E.D.B., and FWO/KAN/1520913N to F.C.) and by IAP project P7/43 (Belspo): Belgian Medical Genomics Initiative. F.C. is a postdoctoral fellow, H.V. and M.B. are doctoral fellows, and B.P.L. and E.D.B. are senior clinical investigators of the FWO.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure S1
(PDF 390 kb)
Supplementary Figure S2
(PDF 331 kb)
Supplementary Table S1
(DOC 339 kb)
Supplementary Table S2
(DOC 322 kb)
Supplementary Table S3
(DOC 353 kb)
Supplementary Table S4
(DOC 322 kb)
Supplementary Table S5
(DOC 318 kb)
Rights and permissions
About this article
Cite this article
Coppieters, F., Van Schil, K., Bauwens, M. et al. Identity-by-descent–guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med 16, 671–680 (2014). https://doi.org/10.1038/gim.2014.24
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.24
Keywords
This article is cited by
-
Clinical features and genetic spectrum of NMNAT1-associated retinal degeneration
Eye (2022)
-
The genetics of rod-cone dystrophy in Arab countries: a systematic review
European Journal of Human Genetics (2021)
-
Identification of a mutation in CNNM4 by whole exome sequencing in an Amish family and functional link between CNNM4 and IQCB1
Molecular Genetics and Genomics (2018)
-
Functional characterization of a novel non-coding mutation “Ghent +49A > G” in the iron-responsive element of L-ferritin causing hereditary hyperferritinaemia-cataract syndrome
Scientific Reports (2017)
-
Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes
Scientific Reports (2017)
