
Abstract
Purpose:
Many regions have implemented newborn screening (NBS) for cystic fibrosis (CF) using a limited panel of cystic fibrosis transmembrane regulator (CFTR) mutations after immunoreactive trypsinogen (IRT) analysis. We sought to assess the feasibility of further improving the screening using next-generation sequencing (NGS) technology.
Methods:
An NGS assay was used to detect 162 CFTR mutations/variants characterized by the CFTR2 project. We used 67 dried blood spots (DBSs) containing 48 distinct CFTR mutations to validate the assay. NGS assay was retrospectively performed on 165 CF screen–positive samples with one CFTR mutation.
Results:
The NGS assay was successfully performed using DNA isolated from DBSs, and it correctly detected all CFTR mutations in the validation. Among 165 screen-positive infants with one CFTR mutation, no additional disease-causing mutation was identified in 151 samples consistent with normal sweat tests. Five infants had a CF-causing mutation that was not included in this panel, and nine with two CF-causing mutations were identified.
Conclusion:
The NGS assay was 100% concordant with traditional methods. Retrospective analysis results indicate an IRT/NGS screening algorithm would enable high sensitivity, better specificity and positive predictive value (PPV). This study lays the foundation for prospective studies and for introducing NGS in NBS laboratories.
Genet Med 18 3, 231–238.
Similar content being viewed by others

Introduction
Cystic fibrosis (CF) is the most common life-threatening autosomal recessive disease among Caucasians, occurring in ~1 in 4,000 newborns, and is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Although over 1,900 CFTR mutations have been reported in the CF Mutation Database (http://www.genet.sickkids.on.ca), it has recently been recognized that the majority of those mutations are not associated with CF.1 There has been a recent effort to rigorously ascertain the disease liability of CFTR mutations through a pivotal project known as CFTR2, the Clinical and Functional Translation of CFTR.1 The CFTR2 project initially found that 159 CFTR variants account for 96.4% of CF alleles in their large-scale meta-analysis,1 and as of 4 April 2014, there are reported 176 CF-causing mutations, 12 that are not CF-causing variants, and 11 mutations with varying clinical consequences; seven variants are still under evaluation (http://www.cftr2.org).
Because of growing evidence of the benefits of early diagnosis for patients with CF,2 many regions worldwide, including every state in the United States, have implemented routine newborn screening (NBS) for CF. Most CF NBS programs use a method of analyzing a limited number of CFTR gene mutations (typically 23–40) following an immunoreactive trypsinogen (IRT) analysis on dried blood spot (DBS) specimens (the two-tier IRT/DNA algorithm).3,4 The sensitivity of the two-tier test is largely determined by the IRT cutoff value chosen, but it is also influenced by the number of mutations in the CFTR panel.5 Infants with a high IRT level and either one or two mutations detected are reported as screen positive and are referred for sweat testing in diagnostic evaluations. In at least 10% of cases, however, the sweat results are inconclusive.6 Moreover, ~90% of infants with one mutation have normal sweat test results and are therefore CF heterozygote carrier infants who are categorized as screening false positive.7 These CF NBS false-positive results can cause parental anxiety,8,9,10 add considerable expense to NBS,11 and may delay the diagnosis of CF when one mutation is detected9 or insufficient quantities of sweat are obtained.6 Therefore, the challenges associated with false positives based on carrier detection in the common IRT/DNA protocol are significant and magnified by the disproportionately large number of carriers in the population with high IRT levels.9,10 Although restricting the number of CFTR mutations in CFTR panels can reduce carrier detection and potentially improve the positive predictive value (PPV), the NBS goals of equity and the highest possible sensitivity become more difficult to achieve. Finally, NBS via the IRT/DNA algorithm has identified a new and unexpected challenge: identifying infants who do not meet the criteria for a diagnosis of CF but are labeled as having CFTR-related metabolic syndrome (CRMS) in the United States since 2009.12,13
In a quality improvement effort intended to address the shortcomings of the current IRT/DNA algorithm and therefore enhance NBS for CF, we undertook a project to strategically evaluate expanded DNA analyses in NBS for CF targeting only CF-causing mutations. The NGS platform was a logical assay because of its capability to simultaneous detect a large number of mutations in a scalable manner with the turnaround time required in a NBS laboratory setting.14 Specifically, we used the investigator-use-only (IUO) version of a CF assay that was designed to detect simultaneously a panel of 162 CFTR mutations/variants for which clinical consequences have been described in the CFTR2 project. Here we report our experience of validating this NGS method using 67 DBS samples with 48 distinct, known CFTR mutations. We assessed the robustness of the assay using a laboratory-developed simple DNA isolation method with DBS specimens to demonstrate the feasibility of introducing NGS into high-throughput NBS laboratories with a fast-paced routine. In addition, in a retrospective study designed to assess the potential applicability of an IRT/NGS algorithm to Wisconsin NBS for CF, we performed this NGS assay on 165 residual NBS-positive specimens that were identified with only one of 23 CFTR mutations recommended by the American College of Medical Genetics and Genomics,15 the panel currently used for NBS in Wisconsin.7
Materials and Methods
Study cohort and DBS specimens
A total of 67 DBS specimens were used to verify and validate the assay: 44 de-identified residual NBS specimens with two CFTR mutations identified through NBS or a clinical confirmatory testing process, and 23 CF proficiency testing samples from the Newborn Screening and Molecular Biology Branch at the Centers for Disease Control and Prevention (CDC).16 Those 67 DBS specimens contained 48 distinct CFTR mutations included in the 162-mutation/variant NGS panel. The status of poly-T and TG repeats in intron nine was known for nine Wisconsin samples and all 23 samples provided by the CDC.
From 1 June 2012 to 31 May 2013, Wisconsin screened 61,257 newborns for CF using the two-tier IRT/DNA algorithm. Specimens within the top 4% of the daily IRT values were tested for the American College of Medical Genetics and Genomics panel of 23 CFTR mutations, and the infants with one or more CFTR mutations detected were considered screen-positive for CF (n = 8 with two CFTR mutations, n = 184 with one CFTR mutation). Among the group with one mutation, 165 specimens had sweat test results and/or clinical assessment available. Those specimens were de-identified and used in this analysis by both the University of Wisconsin and CDC. All NGS assay results were compared with the sweat test results and/or clinical outcomes for concordance. This study was approved by the Health Sciences Institutional Review Board at the University of Wisconsin–Madison as not constituting research as defined under 45 CFR 46.102(d). The CDC’s Office of Science at the National Center for Environmental Health determined the CDC’s involvement as not involving identifiable human subjects under 45 CFR 66.012(d).
DNA isolation
Each DNA sample was isolated from a 3.2-mm DBS punch in Generation DNA Purification Solution and Elution Solution (Qiagen, Valencia, CA) using manufacturer-recommended wash steps with some modifications. DNA then was eluted in 24 µl of molecular-grade water and incubated at 99 °C for 25 minutes.17 DNA also was isolated using a laboratory-developed, one-step method. Each 3.2-mm DBS punch was incubated with 54 µl of DNA elution buffer (5 mmol/l potassium hydroxide, 7.5 mmol/l potassium chloride, and 15 mmol/l Tris-base at 95 °C for 25 minutes. Both methods used 5 µl of isolated DNA for the NGS assay.
NGS assay for detection of CFTR mutations/variants
CFTR mutations are described using both the international nomenclature of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/) and legacy mutation nomenclature (http://www.cftr2.org/browse.php). The assay consisted of the Illumina MiSeqDx IUO CF assay system (Illumina, San Diego, CA), which includes reagents to sequence the full coding region and intron/exon boundaries of the CFTR gene from genomic DNA samples. The MiSeq Reporter data analysis masks sequence data to reveal the status of only the 162 mutations/variants in the CFTR gene characterized by the CFTR2 project, including 127 single-nucleotide substitution mutation/variants, 32 insertion/deletion mutations, exon 2–3 and exon 22–23 deletions, and the poly-TG and TG repeats region in exon 9 ( Table 1 ). In this validation study we included mutations that definitely cause CF, as defined by the CFTR2 project,1 and some that are classified therein as mutations with varying clinical consequences because alleles in the latter category were known to be present in the DBS specimens we analyzed.
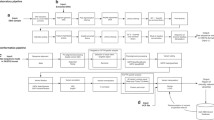
The NGS library to build CFTR gene amplicon libraries was prepared using MiSeqDx IUO CF assay reagents (Illumina) per the manufacturer’s instructions. The process started with the hybridization of CFTR gene targeted oligonucleotides (probes) to unfragmented genomic DNA isolated from DBS. Each probe was also tagged with a common oligonucleotide. DNA templates for polymerase chain reaction (PCR) were formed by extending and ligating 80 targets ranging from 175 to 225 bp DNA fragments. They contained the regions of interest and were flanked by the common oligonucleotide sequences that were partially complementary to PCR primer sequences. Using indexed primers supplied with the IUO kit, the DNA templates then were amplified using PCR to generate the libraries. The indexed primers allowed multiplexing of 48 individual DNA samples. Libraries were normalized with a magnetic bead–based bind–wash–elute protocol before they were pooled into a single tube for sequencing. The sequencing library preparation process is illustrated in Figure 1 .

Sequencing library generation. Each probe contains two parts of sequences: one for targeting interested CFTR gene regions, and another for annealing to polymerase chain reaction (PCR) primers. The DNA templates containing targeted CFTR regions were generated by a sequential process consistent with hybridization, extension, and ligation. The indexes included in PCR primers allow multiplexing of 48 individual DNA samples. The PCR primers also are designed to include P5 and P7, which are necessary for initiating sequencing.
Automated sequencing was accomplished using Illumina’s Sequencing by Synthesis technology,18 and 48 samples were indexed, normalized, pooled, and loaded onto the MiSeqDx reagent cartridge. Data were generated and analyzed with the IUO versions of MiSeq Operating Software and Real Time Analysis software. The base calls were generated by the IUO versions of the MiSeqDx sequencing system, and MiSeq Reporter software performed demultiplexing, alignment, and variant calling. Results from each of the 48 samples were presented in a text file including assay performance (pass or fail), sample call rate (percentage of successful analyses of targeted regions), status of poly-T and poly-TG polymorphisms, detected mutation’s name, type, CFTR gene region, genomic location, and cDNA and protein names according to Human Genome Variation Society nomenclature.
Sanger sequencing
All CF-causing or varying consequence mutations newly identified by the MiSeqDx Cystic Fibrosis System were verified by a traditional Sanger sequencing method performed at the Newborn Screening and Molecular Biology Branch at the CDC in Atlanta, GA. The CFTR gene was amplified and sequenced using primer sets described in the National Center for Biotechnology Information’s Probe Database, a public registry of nucleic acid reagents. The primer sets (RSS000010013) were previously available as a resequencing assay from Applied Biosystems (Grand Island, NY). The RSS000010013 primer sets were used to amplify specific target regions identified by NGS using HotStarTaq Master Mix (Qiagen) with the following cycling conditions: a 10-min denaturing step at 95 °C, followed by 40 cycles at 95 °C for 30 s, 62 °C for 30 s, and 72 °C for 1 min, followed by an extension at 72 °C for 10 min and an indefinite hold at 4 °C. Unused primer and nucleotides were removed using ExoSAP-IT (USB Corp., Cleveland, OH), and sequencing was performed using BigDye Terminator Ready Reaction kit version 1.1 (Life Technologies). Cycle sequencing reactions consisted of 1 μl of PCR product, 1 μl of BigDye Terminator version 1.1, 1.5 μl of 5× sequencing buffer, and 3.2 pmol of primer. Additional primers for regions not covered by the RSS000010013 primer sets were used to characterize the CFTR gene as previously described.19 All sequencing reactions were purified using the BigDye XTerminator and were electrophoresed with run module BDx_Rapid-Seq36_POP7; data were analyzed using SeqScape software (Life Technologies, Grand Island, NY). DNA sequences were aligned with GenBank CFTR genomic reference sequence NG_016465.
Results
DBS DNA isolation and NGS assay performance
Successful NGS assay runs were obtained on DNA samples isolated using both Qiagen and the laboratory-developed methods. A 100% sample call rate was obtained from 67 DBS specimens with 48 distinct known CFTR mutations, and all mutations were 100% concordant with previously identified mutations. In the 32 (of 67) specimens with known intron 9 poly-T and TG repeat status, there was 100% concordance with previously obtained results. The intron 9 poly-T in the tested samples included 5T, 7T, and 9T. The intron 9 TG repeat ranged in size from 9 to 13 repeats.
Concordance analysis
NGS assessment of 165 specimens with one CFTR mutation identified through routine NBS revealed no additional disease-causing mutations in 156 infants; 151 specimens were consistent with their normal sweat chloride test results. In five samples one additional disease-causing mutation was found, and the newborn had either an abnormal sweat chloride test result (sweat chloride > 30 mmol/l) or was diagnosed with CF or CRMS based on additional clinical or genetic evaluations. CFTR mutations with varying consequences were identified in four additional specimens; one had an abnormal sweat chloride test result, whereas the other three had normal sweat chloride test results. Of the four CFTR mutations with varying consequences, two cases with an F508del/D1152H genotype were labeled “carrier” based on the sweat test results and the initial clinical assessment, whereas another F508del/D1152H genotype was labeled as CRMS. Note that the D1152H mutation is associated with variable clinical consequences. Overall, four of these nine infants with a second mutation identified are currently receiving follow-up care as cases of CRMS ( Table 2 ).
No second disease-causing mutation was found in five specimens that had abnormal sweat chloride test results. Three of these five children have been diagnosed with classical CF, and the other two have been diagnosed with CRMS. Mutations were identified in two of the five samples through the confirmatory testing process during NBS follow-up ( Table 3 ).
Comparison of screening validity measures with routine IRT/DNA and NGS
The PPV of the routine Wisconsin IRT/DNA method evaluating 23 American College of Medical Genetics and Genomics–recommended mutations after an IRT analysis was 7.3%. When these 165 one-mutation samples were evaluated with the NGS panel (MiSeqDx IUO 162 mutations/variations), the PPV would have been 77.8% if a positive screen is defined as two CF-causing mutations. However, the sensitivity of the IRT/NGS algorithm would have decreased as much as 50% for classic CF cases when a positive screen is defined as two CF-causing mutations because of uncommon mutations found in five patients with classic CF that are not included in the MiSeqDx IUO 162 mutations/variations panel.
Discussion
In 2013 the 50-year anniversary of NBS was celebrated and many key developments were highlighted, especially those from the past decade. As with most science, advances in technology and methodology have generally driven improved screening procedures in public health laboratories.20 Discovery of the CFTR gene21 allowed for a second-tier mutation-detection algorithm3,4 for CF NBS, which greatly increased the sensitivity of the original IRT algorithms.22,23 Initially, the two-tier IRT/DNA method incorporated only a single, common mutation, F508del (c.1521_1523delCTT), followed later by an expanded panel of mutations (typically 23–40 mutations).7,23 These expanded mutation panels add value by improving the sensitivity of CF screening, but they remain insufficient and sometimes include mutations that are not CF-causing alleles.
Despite its great advantages23,24,25 and popularity, there are still many challenges with IRT/DNA screening, which is now practiced worldwide and is the most widely used algorithm for CF NBS.5 One of the biggest challenges is a lower than ideal PPV resulting in ~10 heterozygote newborn carriers (i.e., a high IRT and single identified CFTR mutation) for every CF case diagnosed after follow-up sweat testing. This is exacerbated by the occurrence of an insufficient quantity of sweat collected (QNS) and/or indeterminate sweat test outcomes in 10–20% of the screen-positive infants. In addition, CFTR panels being used have insufficient mutations to allow the detection of minority populations with uncommon CF-causing mutations that can cause inequities in NBS, such as M1101K (c.3302T>A), more commonly found in Hutterite populations,26 and H199Y (c.595C>T) and S492F (c.1475C>T), more commonly found in Hispanic populations. As the genetic diversity of populations increases, the current panels used for IRT/DNA screening will not adequately meet the need; “missed cases” are more likely among non-Caucasians. In addition, greatly expanded CFTR analysis with gene scanning, as done in the California NBS program,27 rather than targeted mutation analysis with NGS, can lead to the identification of more CRMS cases than CF cases, as well as other inconclusive outcomes. Another shortcoming of currently used CFTR screening panels is that they also fail to detect class III mutations other than G551D (c.1652G>A) that can now be treated effectively by CFTR modulators.28,29
The overall goal of this project was to develop a strategy for improving equity and increasing the chance of identifying two CF-causing mutations in infants with CF through NBS. The project took advantage of NGS technology combined with the increased knowledge about CF-causing mutations determined by the CFTR2 project. The original CFTR mutation database identified and described 1,956 mutations/variants in the CFTR gene (http://www.genet.sickkids.on.ca/cftr/app, accessed 30 April 2014). The CFTR2 project, however, which includes assessments of over 40,000 patients, found that only about 10% are actually CF-causing mutations, providing a major advance in our understanding of CF. Combining this new information with advanced molecular technology has allowed us to study new screening strategies that may improve follow-up processes.
Our experience of performing an NGS assay using DNA isolated from DBS specimens described in this study indicated that those DNA extracts can reliably be used. We showed that DNA of sufficient quality could be routinely isolated from DBS not only using a multistep method but also using a laboratory-developed one-step method. Our laboratory also showed that this simplified one-step DNA isolation method provides sufficient DNA for other PCR-based molecular testing applications including real-time quantitative PCR and Sanger sequencing (data not shown). The NGS method we used proved to be robust and reliable, as evidenced by 100% concordance with the validation of samples representing 48 distinct CFTR mutations. Further evidence of the robustness of this assay was seen in the analysis of the poly-T and TG repeats in intron 9 of CFTR. This complex region contains between 9 and 13 TG repeats followed by a homopolymer T region of 5, 7, or 9T. We obtained the concordant results on all samples for which this region was previously characterized, including samples with two of the same poly-T or TG repeats (i.e., 7T/7T and 11TG/11TG) and two different poly-T or TG repeats (i.e., 5T/7T and 10TG/11TG).
The NGS assay also allowed us to simultaneously assess 46 patient samples, along with a positive and a negative control sample, and to generate auto-call results for the panel of 162 mutations/variants using the provided software. Although this project focused on the validation of NGS in a public health laboratory, our experience suggests that an IRT/NGS algorithm can potentially contribute to improved timeliness because nearly all patients with CF could be identified genetically from the DBS specimens within one week. When two CF-causing mutations are detected, the confirmation of a CF diagnosis by sweat testing can be routinely achieved within two weeks.9
The CFTR2 project estimated that the inclusion of 139 CFTR mutations should encompass 97% of known CF-causing mutations.1 However, our data show that even in a region like Wisconsin, with a modest degree of genetic diversity, there are some patients whose second mutation is not detected by a 139-mutation panel. Because Wisconsin has genotype data on more than 500 patients with CF diagnosed through screening, we sought to understand how many CF cases have uncommon mutations not currently on the CFTR2 list; we found the five cases reported herein as well as other ones found previously, including G1047R, also referred to as 3139 G>C (c.3271G>C), Y849X (c.2679C>A), and 2043delG (c.2043delG). These observations stress the need for a more inclusive CF-causing mutation NGS panel to achieve a higher sensitivity when it is used for NBS. More than 20 additional CF-causing mutations not listed in Table 1 have been added since the initial publication of the CFTR2 project.1 Fortunately, the MiSeqDx CF sequencing assay already generates sequence information that covers these additional CFTR2-listed mutations, and it could be expanded using only bioinformatics changes to reveal more mutations as their disease liability is better understood.
This study provides evidence that a more inclusive CFTR mutation panel has the potential to reduce false-positive results caused by CF heterozygote carrier infants identified using the conventional IRT/DNA screening algorithm. As the CFTR2 list of CF-causing mutations continues to expand, it is conceivable that the definition of a positive screening test might be changed to require the detection of two mutations, particularly if the PPV exceeds 95% with an expanded panel of 200 or more CF-causing mutations detected by NGS. The practical advantages of using such an expanded panel can clearly be seen in this one year assessment comparing traditional IRT/DNA screening in Wisconsin with IRT/NGS, where 151 of 153 false-positive screens could have been avoided. On the other hand, the NGS method we used could be relied on neither to identify “private” mutations nor, given its current technological limitations, to be used to detect all the CFTR gross genomic rearrangements such as the deletions and duplications that have been described in about 1% of patients with CF.30 If the definition of a positive CF NBS test was changed with IRT/NGS to require the detection of two CF-causing mutations, the reporting of results to health care providers would need to describe these limitations and include an alert similar to that provided for ultrahigh IRT levels and a negative DNA tier (sometimes called a “safety net”).
In conclusion, there are many potential advantages of an IRT/NGS screening algorithm. These include earlier confirmation of presumptive diagnosis (facilitating expedited initiation of therapy even before sweat testing and assessment of pancreatic functional status), more equity in diagnosis among ethnic groups that have a lower incidence of F508del and other common mutations, and a potential reduction in the number of sweat tests because of the extremely low risk of CF in infants with one mutation and in those with ultrahigh IRT levels. Moving forward, we are conducting a prospective study using an IRT/NGS model to provide evidence of benefits and to identify any new challenges that IRT/NGS may bring to NBS for CF.
Disclosure
The authors declare no conflict of interest.
References
Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 2013;45:1160–1167.
Balfour-Lynn IM . Newborn screening for cystic fibrosis: evidence for benefit. Arch Dis Child 2008;93:7–10.
Ranieri E, Lewis BD, Gerace RL, et al. Neonatal screening for cystic fibrosis using immunoreactive trypsinogen and direct gene analysis: four years’ experience. BMJ 1994;308:1469–1472.
Gregg RG, Wilfond BS, Farrell PM, Laxova A, Hassemer D, Mischler EH . Application of DNA analysis in a population-screening program for neonatal diagnosis of cystic fibrosis (CF): comparison of screening protocols. Am J Hum Genet 1993;52:616–626.
Clinical and Laboratory Standards Institute (CLSI). Newborn Screening for Cystic Fibrosis: Approved Guideline. CLSI document I/LA35-A. Clinical and Laboratory Standards Institute: Wayne, PA, 2011.
Legrys VA, McColley SA, Li Z, Farrell PM . The need for quality improvement in sweat testing infants after newborn screening for cystic fibrosis. J Pediatr 2010;157:1035–1037.
Baker MW, Groose M, Hoffman G, Rock M, Levy H, Farrell PM . Optimal DNA tier for the IRT/DNA algorithm determined by CFTR mutation results over 14 years of newborn screening. J Cyst Fibros 2011;10:278–281.
Tluczek A, Koscik RL, Farrell PM, Rock MJ . Psychosocial risk associated with newborn screening for cystic fibrosis: parents’ experience while awaiting the sweat-test appointment. Pediatrics 2005;115:1692–1703.
La Pean A, Farrell MH, Eskra KL, Farrell PM . Effects of immediate telephone follow-up with providers on sweat chloride test timing after cystic fibrosis newborn screening identifies a single mutation. J Pediatr 2013;162:522–529.
Castellani C, Picci L, Scarpa M, et al. Cystic fibrosis carriers have higher neonatal immunoreactive trypsinogen values than non-carriers. Am J Med Genet A 2005;135:142–144.
Wells J, Rosenberg M, Hoffman G, Anstead M, Farrell PM . A decision-tree approach to cost comparison of newborn screening strategies for cystic fibrosis. Pediatrics 2012;129:e339–e347.
Cystic Fibrosis Foundation, Borowitz D, Parad RB, et al. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J Pediatr 2009;155:S106–S116.
Ren CL, Desai H, Platt M, Dixon M . Clinical outcomes in infants with cystic fibrosis transmembrane conductance regulator (CFTR) related metabolic syndrome. Pediatr Pulmonol 2011;46:1079–1084.
Metzker ML . Sequencing technologies - the next generation. Nat Rev Genet 2010;11:31–46.
Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 2004;6:387–391.
Earley MC, Laxova A, Farrell PM, et al. Implementation of the first worldwide quality assurance program for cystic fibrosis multiple mutation detection in population-based screening. Clin Chim Acta 2011;412:1376–1381.
Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol 2009;124:522–527.
Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008;456:53–59.
Cordovado SK, Hendrix M, Greene CN, et al. CFTR mutation analysis and haplotype associations in CF patients. Mol Genet Metab 2012;105:249–254.
Guthrie R, Susi A . A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338–343.
Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989;245:1073–1080.
Rock MJ, Mischler EH, Farrell PM, et al. Newborn screening for cystic fibrosis is complicated by age-related decline in immunoreactive trypsinogen levels. Pediatrics 1990;85:1001–1007.
Kloosterboer M, Hoffman G, Rock M, et al. Clarification of laboratory and clinical variables that influence cystic fibrosis newborn screening with initial analysis of immunoreactive trypsinogen. Pediatrics 2009;123:e338–e346.
Comeau AM, Parad RB, Dorkin HL, et al. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: a cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics 2004;113:1573–1581.
Gregg RG, Simantel A, Farrell PM, et al. Newborn screening for cystic fibrosis in Wisconsin: comparison of biochemical and molecular methods. Pediatrics 1997;99:819–824.
Zielenski J, Fujiwara TM, Markiewicz D, et al. Identification of the M1101K mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and complete detection of cystic fibrosis mutations in the Hutterite population. Am J Hum Genet 1993;52:609–615.
Kammesheidt A, Kharrazi M, Graham S, et al. Comprehensive genetic analysis of the cystic fibrosis transmembrane conductance regulator from dried blood specimens–implications for newborn screening. Genet Med 2006;8:557–562.
Van Goor F, Yu H, Burton B, Hoffman BJ . Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 2014;13:29–36.
Ramsey BW, Davies J, McElvaney NG, et al.; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–1672.
Férec C, Casals T, Chuzhanova N, et al. Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur J Hum Genet 2006;14:567–576.
Acknowledgements
This study was funded by the Legacy of Angels Foundation. The authors thank Laurence Lee for his assistance with the figure.
Mention of any company or product does not constitute endorsement by the Centers for Disease Control and Prevention.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Baker, M., Atkins, A., Cordovado, S. et al. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: a technical feasibility study. Genet Med 18, 231–238 (2016). https://doi.org/10.1038/gim.2014.209
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.209
Keywords
This article is cited by
-
Scalable, high quality, whole genome sequencing from archived, newborn, dried blood spots
npj Genomic Medicine (2023)
-
Screening for cystic fibrosis in New York State: considerations for algorithm improvements
European Journal of Pediatrics (2016)
