
Abstract
Purpose:
To evaluate the clinical utility of chromosomal microarrays for prenatal diagnosis by a prospective study of fetuses with abnormalities detected on ultrasound.
Methods:
Patients referred for prenatal diagnosis due to ultrasound anomalies underwent analysis by array comparative genomic hybridization as the first-tier diagnostic test.
Results:
A total of 383 prenatal samples underwent analysis by array comparative genomic hybridization. Array analysis revealed causal imbalances in a total of 9.6% of patients (n = 37). Submicroscopic copy-number variations were detected in 2.6% of patients (n = 10/37), and arrays added valuable information over conventional karyotyping in 3.9% of patients (n = 15/37). We highlight a novel advantage of arrays; a 500-kb paternal insertional translocation is the likely driver of a de novo unbalanced translocation, thus improving recurrence risk calculation in this family. Variants of uncertain significance were revealed in 1.6% of patients (n = 6/383).
Conclusion:
We demonstrate the added value of chromosomal microarrays for prenatal diagnosis in the presence of ultrasound anomalies. We advocate reporting back only copy-number variations with known pathogenic significance. Although this approach might be considered opposite to the ideal of full reproductive autonomy of the parents, we argue why providing all information to parents may result in a false sense of autonomy.
Genet Med 16 6, 469–476.
Similar content being viewed by others
Introduction
Conventional karyotyping has been considered the gold standard for routine prenatal genetic diagnosis for many decades now, allowing for microscopic visualization and inspection of chromosomes and thus detection of numerical and structural chromosomal rearrangements. The main limitations are the resolution achieved by G-banding, which is limited to 5–10 Mb at best, and the requirement for cultured cells, needing a minimum of 8–10 days. The introduction of targeted methods of analysis such as fluorescence in situ hybridization (FISH) and multiplex ligation-dependent probe amplification (MLPA) overcome the time constraints and resolution limitations inherent to karyotyping but do not provide a genome-wide analysis.1 More recently, molecular karyotyping using genomic microarrays has reached mainstream use in the postnatal diagnostic setting, providing a genome-wide screen for genomic imbalances at a far superior resolution to karyotyping.2 A number of studies have demonstrated the feasibility of prenatal diagnosis by genomic arrays using a variety of platforms,3,4,5,6,7 but challenges remain in applying high-resolution genomic arrays to prenatal diagnosis.6,8,9 One of the major ethical issues often raised is how to deal with variants of uncertain significance (VOUS) or risk loci, the detection of which leads to additional challenges for genetic counseling of parents. Furthermore, array analysis may reveal an imbalance for known “risk loci” where the future penetrance is uncertain or may be associated with variable expression. The penetrance risks for a number of recurrent copy-number variations (CNVs) have been estimated based on the frequencies in patients and controls.10,11,12 However, although it is possible to calculate a population-based risk, it is impossible in the prenatal setting to predict the phenotypic outcome in the child.
In this article, we present results of a prospective study on the routine use of array comparative genomic hybridization (aCGH) for fetuses with congenital abnormalities detected prenatally by ultrasound from two Belgian genetics centers. We also discuss our approaches to the interpretation and reporting of genomic array results in the prenatal setting.
Materials and Methods
Referral criteria and time period
Samples were received over a 3-year period at the Centre for Human Genetics, KU Leuven, UZ Leuven, and the Centre for Medical Genetics Ghent, Ghent University Hospital (herein referred to as KUL and UG, respectively). For both the centers, inclusion criteria were either multiple abnormalities or isolated abnormality observed on ultrasound for which invasive testing and genetic analysis is advised. Ethical approval was granted for this study in both centers.
Sample types
Chorionic villi samples (CVSs), amniotic fluid (AF) samples, and fetal blood samples were received. In all cases, backup cultures were initiated for further DNA requirements and for conventional karyotype. When available, genomic DNA was immediately isolated from fresh material (including all fetal blood samples). Where required, DNA was isolated from cultured cells (AF samples and CVSs only). Villi from CVSs were separated from maternal tissue under a microscope to minimize maternal cell contamination (MCC). Two to four villi were provided for DNA extraction. Between 2 and 13 ml (mean = 7; median = 8) of AF samples was provided for DNA extraction. AF samples were centrifuged and cell pellets washed twice in phosphate-buffered saline. Genomic DNA was extracted by both centers using the DNA mini kit (QIAGEN Benelux B.V. - Belgium. Venlo, Netherlands) following the manufacturers recommendations. For CVSs, incubation at 56 °C with proteinase K and tissue lysis buffer (Buffer ATL) was performed for at least 1 hour for efficient digestion and lysis of the complete sample. For AF samples, incubation at 56 °C with proteinase K and lysis buffer (Buffer AL) was performed for 10 minutes. Amicon YM-30 Spin Columns (Merck Millipore S.A./N.V., Overijse, Belgium) were used for concentrating DNA samples (KUL). Total DNA yields for the first 100 samples including uncultured CVSs and AF samples ranged from 1 to 6 µg and from 100 ng to 2 µg (mean = 800 ng), respectively. MCC was assessed on DNA for all CVSs using the DNA Purity Assay (Multiplicom, Niel, Belgium) or the PowerPlex system (Promega Benelux BV, Leiden, Netherlands), both of which use polymorphic short tandem repeat (STR) marker analysis. MCC was assessed on DNA for heavily blood-stained AF samples only.
Genomic array platforms
KUL applied the CytoSure Syndrome Plus 105K array and CytoSure Syndrome Plus 180K array (Oxford Gene Technology, Oxford, UK). This platform has genome-wide coverage with enrichment of target regions. Details of the array designs are available from Oxford Gene Technology (http://www.ogt.co.uk/) or the authors. UG applied the Agilent 60K platform (AMADID 21924) or the Agilent 180K platform (AMADID 27676) (Agilent Technologies S.A./N.V., Diegem, Belgium), which is the International Standards For Cytogenomic Arrays Consortium (ISCA; http://www.iscaconsortium.org) design, supplemented with selected genes/loci of interest. Details of the array designs are available from Agilent Technologies or the authors.
aCGH analysis and interpretation
Genomic DNA was labeled for 4 hours using the CytoSure Labelling Kit (Oxford Gene Technology), with no enzyme digestion. Hybridization was performed from 24 to 60 hours in a rotator oven (SciGene, CA, USA) at 65 °C. Washing of arrays was performed using Agilent wash solutions manually or with a Little Dipper Microarray Processor (SciGene) and dried using acetonitrile. Arrays were scanned using an Agilent microarray scanner at 2-µm resolution, followed by calculation of signal intensities using Feature Extraction software (Agilent Technologies). Visualizations of results and data analysis were performed using the CytoSure Interpret Software (Oxford Gene Technology) and the circular binary segmentation algorithm. The calling thresholds were deviation of a circular binary segmentation (CBS) segment from zero log ratio of +0.36 for duplications and −0.72 for deletions and containing ≥5 oligonucleotide probes. All samples were hybridized twice in dye swap experiments, labeled with Cy5 and Cy3 and hybridized versus Cy3 and Cy5-labeled reference DNA, respectively. The dye swap increases the sensitivity which, in turn, allows a more accurate detection of smaller imbalances, refinement of the breakpoint, and mosaicism. No major discrepancies are observed between hybridizations. Results were then classified with CytoSure Interpret Software (Oxford Gene Technology) and with BENCH software (Cartagenia N.V., Leuven, Belgium). Quality control metrics are monitored with CytoSure Interpret software (Oxford Gene Technology). For UG, arrays were hybridized and analyzed as previously described.13 Genomic coordinates are based on build hg18.
Pretest counseling and informed consent
Following detection of an abnormality on ultrasound investigation, array analysis was offered as the first-tier test with additional multidisciplinary counseling and informed consent at a tertiary center. Traditional chromosome analysis by conventional karyotyping was not routinely performed. Rapid aneuploidy detection was performed by FISH or quantitative fluorescent polymerase chain reaction for all patients before array analysis to timely exclude common autosomal and sex chromosome aneuploidies, as well as to exclude triploidy. Parental samples may be required for full interpretation of array results and were provided along with the prenatal sample where possible. Parental samples were only analyzed after the detection of a prenatal imbalance and only for those regions of interest. Patients agree during the informed consent that all causal and clinically significant imbalances, but not all VOUS or (inherited) likely benign CNVs, are reported (routinely). These data are retained by the laboratory and available on request. VOUS were defined as CNVs which have not been described before in the literature as pathogenic nor contain genes known to cause haploinsufficiency syndromes but are likely deleterious, based on the functional evidence known about the genes in the CNVs. The published recommendations and guidelines for classification and interpretation of CNVs for constitutional postnatal genetic diagnosis, including VOUS, form the basis for our CNV classification in the prenatal setting also.8,14,15,16 CNVs without genes or known to be common in the population are considered benign. A list of common polymorphic CNVs is curated by the laboratory based on in-house data and previous experience, supported by additional evidence from the database of genomic variants, DECIPHER, and ISCA databases (see Supplementary Table S2 online). A clinical geneticist assists the laboratory in the final decision of CNV classification and advises which imbalances to report to the referring clinician and patient in challenging cases.
Confirmation of findings
All pathogenic CNVs were confirmed by conventional karyotype, FISH analysis, MLPA, and/or quantitative fluorescent polymerase chain reaction.
Results
A combined total of 403 prenatal samples underwent analysis by aCGH. Of these 403 samples, an interpretable result was obtained in 383 cases. For 20 samples, the array result could not be interpreted due to either MCC (n = 4) or a poor-quality array result with a derivative log ratio (DLR) value beyond an acceptable range of 0.3 (n = 16). Of the total samples received, 65% (n = 262) were AF samples, 21% (n = 85) were CVSs, and 14% (n = 56) were fetal blood samples. Only common polymorphic and likely benign CNVs were observed for 289 of 383 patients (75.5%), and a normal array result was reported. In the remaining 24.5% of patients (n = 94), rare and/or likely pathogenic CNVs were detected, which are further detailed below. For 51 of 383 patients (13.3%), the findings were classified as rare inherited and likely benign CNVs following parental array analysis. The information about those variants was deemed not of clear “clinical relevance” based on current knowledge and hence was not reported back to parents. However, it cannot be excluded that such inherited variants could be pathogenic in the offspring due to incomplete penetrance or due to variable expressivity (for the rationale, see Discussion). Array analysis revealed causal imbalances in 37 of 383 patients (9.6%), 10 of which (2.6%) were submicroscopic. This means approximately one-fourth of all causal imbalances are submicroscopic and thus undetectable by conventional karyotype alone. In 3.1% (n = 12) of patients with abnormal array results (30% of causal imbalances), concordance with the subsequent abnormal karyotype was observed. For 1.6% (n = 6) of patients, VOUS were detected, and the decision of whether to report these VOUS or not was determined on a case-by-case basis, and the rationale is described further below.
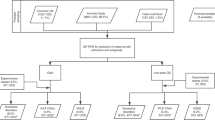
The workflow shown in Figure 1 summarizes our results, with further details of causal imbalances revealed by array analysis, as well as incidental findings of clinical significance and VOUS provided in Supplementary Table S1 online). Notable examples are discussed in more detail below.

Workflow of chromosomal microarray results. CNV, copy-number variations; MCC, maternal cell contamination; VOUS, variants of uncertain significance.
Submicroscopic CNVs
A 6-Mb deletion of 1p36 was observed in the fetus KUL 2 with an Ebstein malformation and cerebral ventriculomegaly. Of note, 1p36 deletions have occasionally been associated with this rare malformation, and this case adds further evidence of a link between this rare heart abnormality and haploinsufficiency of one or more genes at 1p36.17,18 In the fetus KUL 4, referred due to semi-lobar holoprosencephaly, a novel 2-Mb de novo deletion of 10q24.31–q24.32 was observed. Within the deleted region, several genes are located. However, we consider SUFU haploinsufficiency as the most likely cause for this phenotype because SUFU is a negative regulator of hedgehog signaling.19,20 Genetic variants in this signaling pathway, including in SHH (OMIM no. 142945) and GLI2 (OMIM no. 610829), are linked to holoprosencephaly in humans.
Arrays reveal additional information above karyotype
In 15 of 383 fetuses (3.9%), array analysis revealed additional information above karyotyping alone. These are listed in Supplementary Table S1 online and include inverted deletion duplications (inv del dups), unbalanced translocations, and marker chromosomes. One case of interest was fetus KUL 17 for which deletion of 21q22.3, amplification (i.e., four or more copies) of 21q22.2–q22.3, and duplication of 21q21.3q22.2 were observed on both uncultured and cultured CVS material ( Figure 2 ). The duplicated region displays a reduced dynamic range (i.e., reduced deviation of log2 signal intensity ratios from zero, as opposed to the theoretical deviation of +0.58, and the experimentally observed deviations of +0.45 to +0.55), suggestive of mosaicism. MCC was excluded by STR marker analysis of fetal and parental material. Confined placental mosaicism could not be fully excluded because no AF, fetal blood, or fetal skin fibroblast sample was obtained. The initial rapid aneuploidy testing by FISH analysis with a centromeric probe for chromosome 21 revealed three signals, and the subsequent karyotype revealed one normal and one abnormal (possible ring) chromosome 21. This finding represents a mosaicism with different cell lines that have undergone different numbers of breakage–fusion–bridge cycles,21 which has, to our knowledge, not been described previously in a constitutional disorder.

A complex rearrangement of chromosome 21. Duplication of 21q21.3q22.2, amplification of 21q22.2-q22.3, and terminal deletion of 21q22.3 are visible. Oligonucleotide probes are plotted by genomic position on the x-axis and by normalized log2 signal intensity ratios on the y-axis. The duplicated and deleted regions are highlighted, with genes displayed in the lower track and chromosome 21 shown at top of the image.
In fetus KUL 15 referred due to cardiac anomalies, a 17.5-Mb duplication of 22q12.3–q13.33 was detected ( Figure 3a ). Subsequent prenatal karyotype revealed translocation of the duplicated 22q fragment to the 22p arm ( Figure 3c ). Parental karyotypes were both normal. However, parental array analysis revealed a 500-kb duplication of 22q12.3 in the paternal sample ( Figure 3b ). This 500-kb region overlaps with the proximal region of the terminal 22q duplication present in the fetus. We hypothesized this 500-kb region to be an insertional translocation within the 22p microsatellite, which was confirmed by subsequent FISH analysis of the paternal sample (probe RP11-413J08) ( Figure 3d ). The translocation was thus most likely due to a crossing over in the insertional translocation in the father. This may be the first direct support that a submicroscopic insertional translocation can generate a quadrivalent and thus be the cause of a chromosomal rearrangement following recombination.

Paternal insertional translocation as driver of chromosomal translocation. (a) A 17.5-Mb duplication of 22q12.3-q13.33 in the fetus. (b) A 500-kb duplication of 22q12.3 in the paternal sample. Oligonucleotide probes are plotted by genomic position on the x-axis and by normalized log2 signal intensity ratios on the y-axis. The duplicated region is highlighted in (a) and (b), with genes displayed in the lower track and chromosome 22 shown at top of the image. (c) Displays the fetal karyotype with the derivative chromosome 22 (labeled with arrow). (d) Displays the FISH result in the paternal sample, using labeled BAC Clone RP11-413J08, showing three signals, with the additional signal on the derivative chromosome 22.
Detection of mosaicism
In 6 of 383 patients (1.6%), we observe reduced dynamic range (signal intensity ratios with reduced deviation from log2 ratio of zero) for duplicated and deleted regions. These are listed in Supplementary Table S1 online along with details of the FISH (uncultured) and/or karyotype (cultured) confirmatory results. We also observe complete discordance between different fetal samples analyzed. For fetus KUL 28, referred due to heart abnormalities, we received both an amniocentesis and fetal blood sample. A large amplification of the 16q arm was observed by array analysis on fetal blood but not in AF samples (both from DNA of uncultured cells). Karyotyping subsequently revealed an additional marker chromosome 16 in fetal blood but not in cultured amniocytes. Hence, this is a true fetal mosaicism for a marker chromosome 16q. Of note, this finding would not have been identified if AF alone was received. In fetus G 2, a male fetus referred due to increased nuchal translucency, an intragenic 50-kb deletion of the DMD gene was observed with reduced dynamic range, confirmed as mosaic by MLPA analysis on the same CVS DNA. Subsequent analysis by aCGH and MLPA on both uncultured and cultured AF samples could not detect this microdeletion. Coincidentally, the conventional karyotype on both cultured CVSs and AF samples revealed a balanced reciprocal translocation 46,XY,t(3;20)(q27;q11.2) inherited from the father. This translocation apparently has no direct phenotypic consequences, and no deletion of material was observed at the breakpoint by array analysis. aCGH and MLPA performed postnatally on DNA from neonatal blood revealed no evidence of this microdeletion in the dystrophin gene. Hence, this represents a confined placental mosaicism for a DMD microdeletion.
Array analysis of apparently balanced rearrangements
In two familial cases of apparently balanced complex rearrangements, deletions are detected by array analysis at one or more of the breakpoints.
Variants of uncertain significance
VOUS were observed in 6 of 383 fetuses (1.6%). These are listed in Supplementary Table S1 online and consist of both inherited and de novo imbalances. In fetus KUL 37, the 2-Mb deletion of 4q32 was shown to be inherited from the father. The same deletion was previously observed in an affected sibling. However, this child also had a de novo 17q deletion that was considered pathogenic. The deleted region on 4q32 harbors the TLL1 gene, in which heterozygous mutations have been associated with atrial septal defects,22 and large deletions of 4q have also been reported in association with congenital heart defects.23 Because the father was already aware of this deletion, the variant was reported. For fetus G 7, the 2.6-Mb deletion of 18q12.3, although relatively large and de novo, harbors only a single gene (PIK3C3). Similar sized deletions have not been observed in normal individuals or patients. The PIK3C3 gene is a candidate for schizophrenia, but its relation with the ultrasound findings in this fetus remains unclear. PIK3C3 is known to regulate various intracellular membrane-trafficking events. Zhou et al.24 observed that PIK3C3 is required for early embryogenesis and cell proliferation in mice. Because this CNV was de novo, this was reported. In fetus KUL 34, we detected a 434-kb deletion of 15q11.2 in the PWAS BP1/BP2 region, which was subsequently detected in the unaffected mother following parental array analysis. Deletions of this region have been associated with developmental delay and behavioral problems and also other features such as schizophrenia.25,26 Strong phenotypic variability and reduced penetrance are characteristics of deletions of this region, which make predictions of the future phenotype impossible. The most recent and largest study of Rosenfeld et al.12 reports deletions of this region as having a penetrance risk of 10.4% (from 203/25,113 cases (0.81%) and 84/22,246 controls (0.38%)). This CNV was not reported back to the patient based on the lack of any family history of neurodevelopmental or psychological abnormalities, the low penetrance risk, and a lack of evidence for a link to the increased nuchal translucency observed and future penetrance of any phenotypic features. KUL33 was reported because this occurred de novo and contains the BRWD3 gene. Mutations in BRWD3 (OMIM*300553) are associated with X-linked mental retardation and macrocephaly. Four copies of 2q21.3 were detected in KUL35; however, no parental DNA was available. Only six genes are within the region, and hence, this CNV was not reported. For KUL36, the referral reason of death in utero influenced the decision to report this VOUS with no known OMIM genes to the parents.
Discussion
Our findings demonstrate a 2.6% (1.0–4.2%, 95% confidence interval) increase in the diagnostic yield of causal genomic imbalances by discovery of pathogenic submicroscopic CNVs undetectable by conventional karyotype. Novel CNVs and known microdeletion and microduplication syndromes were observed. Our results are slightly lower than those observed in a recent meta-analysis that found a 10% increased diagnostic yield of chromosomal microarrays over karyotyping (95% confidence interval: 8–13%) in the presence of ultrasound anomalies.27 Several recent large-scale prospective studies found rates between 6 and 8% in the presence of ultrasound anomalies.6,7,28 However, we demonstrate that arrays provide additional information over karyotyping in 3.9% of patients. Chromosomal microarrays: (i) provide more precise delineation of deletion and duplication breakpoints for structural rearrangements, allowing for a more accurate assessment of gene content; (ii) accurately identify the origin of additional (euchromatic) chromosomal material; and (iii) can reveal pathogenic imbalances at the rearrangement breakpoints in apparently balanced rearrangements. Combining these findings gives a diagnostic yield of 6.5% above karyotyping alone, which is in line with other published studies. We also show that the identification and localization of CNVs can help in understanding the mechanism causing certain chromosomal rearrangements. This is important to estimate the recurrence risk for future pregnancies. A submicroscopic paternal CNV is an insertional translocation that confers a significant risk to future pregnancies and would have gone unnoticed by conventional karyotype alone. This highlights some previously reported and novel unexpected advantages of array analysis for the investigation of apparently balanced rearrangements.
We observed VOUS in 1.6% of patients, which included both inherited and de novo imbalances. Rates of VOUS ranged from 0.39 to 4.2% depending on whether de novo imbalances were included or not.6,7,27,28 The risk of detecting a pathogenic CNV in the absence of any ultrasound anomalies has been estimated to be between 0.5 and 1.7% in several studies that have included the use of chromosomal microarrays for general screening.28,29,30 Hillman et al.27 comment in their meta-analysis the high degree of heterogeneity in results of different array studies, which may be due to a number of factors including the type of cohort studied and the type of platform used.
We demonstrate the ability to detect mosaicism of varying degrees and size by array analysis in 1.5% of patients, an important requirement if arrays are to be used in place of karyotype. Arrays provide an advantage over conventional karyotyping in the ability to measure mosaicism in DNA from uncultured material, providing an assessment of the level of mosaicism not influenced by the cell culture process. A recent study found mosaicism in a total of 1.2% of patients (n = 43/3,710) investigated by array and karyotype.31 Mosaicism was observed by karyotype alone in 39% of those mosaic cases (n = 17/43); 11 of which showed very high levels (>80% mosaicism) and an abnormal array result, and 6 of which showed a normal array result due to very low levels (<10% mosaicism); 12% of those mosaic cases (n = 5/43) were detected by array alone. Apparently balanced rearrangements were detected in 0.8% of cases (n = 30/3,710).31 However, only ˜6% of de novo apparently balanced rearrangements are considered to be pathogenic,32 and studies have shown that ˜40% of apparently balanced rearrangements harbor pathogenic CNVs at the breakpoints, or elsewhere.33,34 Taken together, it is clear that karyotyping and FISH remain essential tools for accurate prenatal genetic diagnosis, and parental cytogenetic analysis may also be required to achieve a correct diagnosis of recurrence risk as demonstrated by our results and others. Karyotype is required for any family with a known family history of a balanced rearrangement only identifiable by conventional karyotype. In addition, those families with a previous abnormal pregnancy/child for which conventional karyotyping was not performed should also be eligible for karyotyping in order to exclude an unidentified balanced rearrangement as the cause.
Approach to genetic counseling
Given that prenatal screening should be aimed at providing pregnant women with opportunities for meaningful reproductive choices, it is important to determine which types of information regarding CNVs may expand or otherwise even undermine these opportunities. Three main approaches to the reporting of information revealed by array analysis can be discerned. A first approach is to give patients the option during the pretest counseling and informed consent process of which types of information they do or do not wish to be informed of following the analysis.35 A second approach is to reveal all information including the detection of VOUS. In a third approach, only information relating to the purpose of the analysis is provided (i.e., causal and/or clinically significant findings), and information deemed not of clear “clinical relevance” based on current knowledge is not reported to the patient (e.g., detection of VOUS and inherited likely benign CNVs), who agrees to this during the pretest counseling and informed consent process. This third approach is that adopted by the groups involved in this study. All three approaches require thorough pretest counseling.9,36,37,38
In recent years, case–control studies have enabled the quantification of the penetrance risk for certain recurrent microdeletions and microduplications, such as the 16p11.2 deletion and reciprocal duplication.10,11,12 Our approach is that those CNVs with penetrance risk factors below 25% were not reported back routinely, unless deemed to be clinically actionable. Examples of these situations include the following: the 15q11.2 deletion (NIPA1 gene), with penetrance risk 10.4% that was detected in KUL 34 and not reported and the 22q11.2 duplication (TBX1 gene), with penetrance risk 21.9% that was detected in KUL 7 in association with cardiac anomalies. In the absence of cardiac anomalies on the clinical referral, this would nevertheless be reported, and follow-up with detailed ultrasound examination for the presence of cardiac defects would be advised. The main difference between our approach and the laboratories providing all information revealed by arrays to the pregnant women is that the latter will report back rare/novel large (typically more than 500 kb) inherited CNVs and imbalances for known “risk loci” where the future penetrance is uncertain and with low odds. Of note, in Mikhaelian et al.’s39 survey of prenatal genetic counselors in the United States and Canada, the most prevalent ethical issue was the potential for ambiguous results, with 69% (n = 111) of respondents expressing concerns, including about their ability to accurately interpret such results, the possibility a patient may terminate a pregnancy because of such results, and that such results may lead to psychosocial “harm” to a patient.
Conclusion
Our results add support to other recent studies for the use of genomic arrays as the first-tier test for prenatal diagnosis of ultrasound abnormalities, as evidenced by the increased diagnostic yield of pathogenic submicroscopic imbalances and the additional information and faster reporting time offered in comparison to conventional karyotype. Considering the increased diagnostic yield in case of increased risk but in the absence of abnormal ultrasound, the use of chromosomal arrays as the first-tier test for all invasive prenatal referrals seems warranted. Nevertheless, the workflow for classification of CNVs, the subsequent interpretation, and reporting back of results warrants further societal research to determine the best provision of care.
Disclosure
J.R.V. is a consultant of Oxford Gene Technology and the founder of Cartagenia. The remaining authors declare no conflict of interest.
References
Goumy C, Gouas L, Pebrel-Richard C, et al. Prenatal detection of cryptic rearrangements by multiplex ligation probe amplification in fetuses with ultrasound abnormalities. Genet Med 2010;12:376–380.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764.
Coppinger J, Alliman S, Lamb AN, Torchia BS, Bejjani BA, Shaffer LG . Whole-genome microarray analysis in prenatal specimens identifies clinically significant chromosome alterations without increase in results of unclear significance compared to targeted microarray. Prenat Diagn 2009;29:1156–1166.
Faas BH, van der Burgt I, Kooper AJ, et al. Identification of clinically significant, submicroscopic chromosome alterations and UPD in fetuses with ultrasound anomalies using genome-wide 250k SNP array analysis. J Med Genet 2010;47:586–594.
Srebniak M, Boter M, Oudesluijs G, et al. Application of SNP array for rapid prenatal diagnosis: implementation, genetic counselling and diagnostic flow. Eur J Hum Genet 2011;19:1230–1237.
Breman A, Pursley AN, Hixson P, et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat Diagn 2012;32:351–361.
Shaffer LG, Dabell MP, Fisher AJ, et al. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat Diagn 2012;32:976–985.
Vetro A, Bouman K, Hastings R, et al. The introduction of arrays in prenatal diagnosis: a special challenge. Hum Mutat 2012;33:923–929.
Wapner RJ, Driscoll DA, Simpson JL . Integration of microarray technology into prenatal diagnosis: counselling issues generated during the NICHD clinical trial. Prenat Diagn 2012;32:396–400.
Kaminsky EB, Kaul V, Paschall J, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 2011;13:777–784.
Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–846.
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG . Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med 2013;15:478–481.
Buysse K, Delle Chiaie B, Van Coster R, et al. Challenges for CNV interpretation in clinical molecular karyotyping: lessons learned from a 1001 sample experience. Eur J Med Genet 2009;52:398–403.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST ; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–685.
Vermeesch JR, Brady PD, Sanlaville D, Kok K, Hastings RJ . Genome-wide arrays: quality criteria and platforms to be used in routine diagnostics. Hum Mutat 2012;33:906–915.
de Leeuw N, Dijkhuizen T, Hehir-Kwa JY, et al. Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat 2012; e-pub ahead of print 14 February 2012.
Witters I, Vermeesch JR, Gyselaers W, Fryns JP . Distinct prenatal ultrasonographic cranial and cerebral findings in terminal deletion of the short arm of chromosome 1. Prenat Diagn 2002;22:831–834.
Digilio MC, Bernardini L, Lepri F, et al. Ebstein anomaly: genetic heterogeneity and association with microdeletions 1p36 and 8p23.1. Am J Med Genet A 2011;155A:2196–2202.
Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R . The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 2010;24:670–682.
Kim JJ, Gill PS, Rotin L, et al. Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J Neurosci 2011;31:1825–1836.
Voet T, Vanneste E, Van der Aa N, et al. Breakage-fusion-bridge cycles leading to inv dup del occur in human cleavage stage embryos. Hum Mutat 2011;32:783–793.
Stanczak P, Witecka J, Szydlo A, et al. Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD. Eur J Hum Genet 2009;17:344–351.
Xu W, Ahmad A, Dagenais S, Iyer RK, Innis JW . Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32.2-q34.3. Am J Med Genet A 2012;158A:635–640.
Zhou X, Takatoh J, Wang F . The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS ONE 2011;6:e16358.
Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CA, et al. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur J Med Genet 2009;52:108–115.
Sahoo T, Theisen A, Rosenfeld JA, et al. Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med 2011;13:868–880.
Hillman SC, McMullan DJ, Hall G, et al. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol 2013;41:610–620.
Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–2184.
Armengol L, Nevado J, Serra-Juhé C, et al. Clinical utility of chromosomal microarray analysis in invasive prenatal diagnosis. Hum Genet 2012;131:513–523.
Lee CN, Lin SY, Lin CH, et al. Clinical utility of array comparative genomic hybridisation for prenatal diagnosis: a cohort study of 3171 pregnancies. BJOG 2012 119:614–625..
Bi W, Borgan C, Pursley AN, et al. Comparison of chromosome analysis and chromosomal microarray analysis: what is the value of chromosome analysis in today’s genomic array era? Genet Med 2013;15:450–457.
Warburton D . De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 1991;49:995–1013.
De Gregori M, Ciccone R, Magini P, et al. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: a study of 59 patients. J Med Genet 2007;44:750–762.
Schluth-Bolard C, Delobel B, Sanlaville D, et al. Cryptic genomic imbalances in de novo and inherited apparently balanced chromosomal rearrangements: array CGH study of 47 unrelated cases. Eur J Med Genet 2009;52:291–296.
Siegal G, Bonnie RJ, Appelbaum PS . Personalized disclosure by information-on-demand: attending to patients’ needs in the informed consent process. J Law Med Ethics 2012;40:359–367.
de Jong A, Dondorp WJ, Frints SG, de Die-Smulders CE, de Wert GM . Advances in prenatal screening: the ethical dimension. Nat Rev Genet 2011;12:657–663.
Dondorp W, Sikkema-Raddatz B, de Die-Smulders C, de Wert G . Arrays in postnatal and prenatal diagnosis: an exploration of the ethics of consent. Hum Mutat 2012;33:916–922.
McGillivray G, Rosenfeld JA, McKinlay Gardner RJ, Gillam LH . Genetic counselling and ethical issues with chromosome microarray analysis in prenatal testing. Prenat Diagn 2012;32:389–395.
Mikhaelian M, Veach PM, MacFarlane I, LeRoy BS, Bower M . Prenatal chromosomal microarray analysis: a survey of prenatal genetic counselors’ experiences and attitudes. Prenat Diagn 2013;33:371–377.
Acknowledgements
This work has been made possible by the Agency for Innovation by Science and Technology (IWT) (SBO-60848 to J.R.V.); Research Foundation Flanders (FWO) (FWO grant G.0320.07. to J.V.); and University of Leuven (KU Leuven) SymBioSys (PFV/10/016 and GOA/12/015 to J.R.V. and K.D.).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 120 kb)
Supplementary Table S2
(DOC 352 kb)
Rights and permissions
About this article
Cite this article
Brady, P., Delle Chiaie, B., Christenhusz, G. et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genet Med 16, 469–476 (2014). https://doi.org/10.1038/gim.2013.168
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.168
Keywords
This article is cited by
-
Does isolated nuchal translucency from 2.5 to 2.9 mm increase the risk of fetal chromosome disease?
Molecular Genetics and Genomics (2022)
-
A Chinese multicenter retrospective study of isolated increased nuchal translucency associated chromosome anomaly and prenatal diagnostic suggestions
Scientific Reports (2021)
-
Fetoscopic endoluminal tracheal occlusion and reestablishment of fetal airways for congenital diaphragmatic hernia
Gynecological Surgery (2018)
-
Pregnant Genetic Counselors in an Era of Advanced Genomic Tests: What Do the Experts Test Prenatally?
Journal of Genetic Counseling (2018)
-
Importance of complete phenotyping in prenatal whole exome sequencing
Human Genetics (2018)
