
Abstract
Purpose:
The mitochondrial genome is highly polymorphic. A unique feature of deleterious mitochondrial DNA (mtDNA) mutations is heteroplasmy. Genetic background and variable penetrance also play roles in the pathogenicity for a mtDNA variant. Clinicians are increasingly interested in requesting mtDNA testing. However, interpretation of uncharacterized mtDNA variants is a great challenge. We suggest a stepwise interpretation procedure for clinical service.
Methods:
We describe the algorithms used to interpret novel and rare mtDNA variants. mtDNA databases and in silico predictive algorithms are used to evaluate the pathogenic potential of novel and/or rare mtDNA variants.
Results:
mtDNA variants can be classified into three categories: benign variants, unclassified variants, and deleterious mutations based on database search and in silico prediction. Targeted DNA sequence analysis of matrilineal relatives, heteroplasmy quantification, and functional studies are useful to classify mtDNA variants.
Conclusion:
Clinical significance of a novel or rare variant is critical in the diagnosis of the disease and counseling of the family. Based on the results from clinical, biochemical, and molecular genetic studies of multiple family members, proper interpretation of mtDNA variants is important for clinical laboratories and for patient care.
Introduction
Mitochondrial diseases are a group of disorders caused by defects in components of the respiratory chain and are associated with mutations in either mitochondrial DNA (mtDNA) or nuclear genes. Confirming a diagnosis of a mitochondrial disease based on defects in mtDNA is challenging because of the clinical and genetic heterogeneity of the disease. Unlike the disomic nature of most nuclear genes, mtDNA copy number can vary from hundreds to several thousands in a cell. Sequence variants in mtDNA can be present in each mtDNA copy, a property termed homoplasmy, or may occur in a subpopulation of mtDNA copies, referred to as heteroplasmy. Pathogenic mtDNA mutations are often heteroplasmic. The percentage of mutation heteroplasmy can vary among different tissues, thus contributing to the high variability in clinical features and disease severity. In addition, mtDNA is highly polymorphic; mtDNA variants have been reported to occur in almost every nucleotide position of the 16,569-bp mitochondrial genome.1,2 Some variants may contribute to mitochondrial functional defects, as well as underlying susceptibility to various diseases, or can act synergistically with other pathologic variants, but by themselves do not cause disease, and thus are referred to as secondary mutations. Consequently, rare and novel variants must be assessed for their pathogenic potential. Interpretation of these variants thus becomes a great challenge.
In 2008, the American College of Medicine published “Recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007.”3 These recommendations have been widely accepted and used as technical standards and practical guidelines by clinical laboratories. However, these guidelines are primarily developed for the interpretation of nuclear gene variations and do not take into account the concept of heteroplasmy as a variable in the interpretation of pathogenicity. Sequence analysis of the whole mitochondrial genome is now routinely performed as part of the molecular diagnosis of maternally inherited mitochondrial disorders in an increasing number of Clinical Laboratory Improvement Amendment (CLIA)/College of American Pathologists (CAP)-certified laboratories. Therefore, it is important and necessary to have a uniform and practical guideline for the interpretation of mitochondrial DNA (mtDNA) variants. Laboratories that offer mtDNA testing should strive for consensus in how to report mtDNA variants and recommendations for follow-up studies. Here we provide the current algorithms that are used in our laboratory to interpret rare and novel mtDNA variants based on our 20 years of experience sequencing thousands of mitochondrial genomes.
Typically, every person harbors an average of 30 mtDNA variants when compared with the Cambridge reference sequence (NC_012920).4 Depending on ethnic variation that was not taken into account when the reference sequence was developed, some individuals may have over 100 variants. Based in part on their frequent occurrence in subpopulations, a majority of these variants are considered benign. However, any given individual can harbor up to five or more rare and/or novel variants, which can be either homoplasmic or heteroplasmic and may or may not be associated with any disease phenotypes. A single such variant may be the determining factor in disease development.5,6 Although most reported common recurrent pathogenic mutations are heteroplasmic, the rare variants associated with common complex diseases such as Leber hereditary optic neuropathy (LHON), diabetes, and hypertension are often homoplasmic.7 Thus, correctly interpreting the possible pathogenicity rare and novel mtDNA variants is essential in elucidating their clinical significance. An mtDNA variant can be classified as deleterious if the mitochondria harboring the variant can be demonstrated to be deficient in respiratory chain function compared with normal controls in vivo or in vitro. This is often studied through the use of transmitochondrial cybrids.8 However, functional studies are time-consuming, and it is not practical or even possible to perform in vitro or in vivo experiments for every rare variant. Although not always reliable, publically available mtDNA databases and algorithms that examine protein structure/function/evolution can be utilized to gauge the pathogenic potential of novel and/or rare mtDNA variants.
Databases for mtDNA Variants and Mutations
MITOMAP (http://www.MITOMAP.org/MITOMAP)2 compiles human mtDNA variations from both published and unpublished sources. All entries are carefully curated by MITOMAP. The MITOMAP server is currently maintained at The Children’s Hospital of Philadelphia and is updated frequently. All mtDNA variants are classified into two categories in MITOMAP: polymorphisms and mutations. The polymorphism category includes benign polymorphisms, somatic mutations, and collections of unpublished variants. The mutation category contains confirmed mtDNA mutations and variants reported to have disease association. Relevant publications are listed for identified variants.
The Human Mitochondrial Genome Database (mtDB) (http://www.mtdb.igp.uu.se/)1 is another resource with extensive documentation of human mitochondrial variants. It contains mtDNA variants from over 2,700 individuals who were healthy at the time of ascertainment and their frequency in the subject cohort, which is very helpful in interpreting the nature of an mtDNA variant. However, it should be recognized that some variants may appear to be rare because of ethnic underrepresentation in the database.1
The Mamit-tRNA database (http://mamit-trna.u-strasbg.fr/human.asp)9 contains mammalian mitochondrial tRNAs with an emphasis on the structural characteristics. It provides extensive documentation of polymorphisms and mutations in mitochondrial tRNA genes related to human mitochondrial disorders and 2D cloverleaf representations of tRNAs, mutations associated with disease phenotypes, and corresponding references.9
Allele Frequencies of mtDNA Variants
The allele frequencies are obtained from the mtDB database and our private database (comprising mitochondrial whole-genome sequencing for over 3,000 individuals and 420 matrilineal relatives as of June 2011). We use an allele frequency of ≤0.2% as the definition of rare mtDNA variants. The cut-off of 0.2% is arbitrary. Given the fact that most confirmed mtDNA mutations are rare, they are either not being detected in the cohort of 2,704 individuals in mtDB or are detected at very low frequency (2,703:1 = 0.04%). The 0.2% cut-off will not classify many benign variants into the unclassified category and at the same time will reduce the number of likely pathogenic rare variants misclassified as benign variants.
Haplogroups
Because of their unique population history, specific mtDNA haplogroups are identified in different geographic regions. Variant frequencies are different in different haplogroups. Our laboratory has haplogroup data from over 2,000 individuals. When identifying one or more rare variants from one individual, check which haplogroup the individual belongs to. If the rare variants are commonly seen in the same haplogroup, then the low allele frequencies could be caused by ethnic underrepresentation in the database; therefore, these variants are likely benign.
Considerable caution is needed in using data derived from diagnostic laboratories in that almost all samples are obtained from unhealthy individuals. Similarly, there are errors and inconsistency in the medical literature concerning the classification of rare population-specific variants and pathologic alterations.
Additional Resources
Other resources should be used to further determine whether an mtDNA variant has been previously reported with a disease association:
-
1
PubMed (http://www.ncbi.nlm.nih.gov/PubMed/) is an NCBI database for archiving MEDLINE database of references on life sciences.
-
2
Online Mendelian Inheritance in Man (OMIM, http://omim.org/ or http://www.ncbi.nlm.nih.gov/omim) is a comprehensive, authoritative compendium of human genes and clinical phenotypes that is freely available and updated daily. It contains all known Mendelian disorders and over 12,000 genes, as well as selected alleles.
-
3
Google is a powerful Internet search engine useful for a specific mtDNA variant by standard and alternate nomenclatures.10Search by the key word “mtDNA” and all possible ways to denote a variant to retrieve published literature for a certain variant.
Classification of mtDNA Variants
We report the algorithms established and routinely used in our laboratory for the interpretation of mtDNA variants. In general, mtDNA variants can be classified into three categories ( Figure 1 ):
-
1. Benign variant: If a variant has been reported in MITOMAP as a polymorphism, has no report of disease association in the population or family studies, and has been reported in mtDB at a frequency greater than 0.2%, then this variant is considered likely a benign variant
-
2. Unclassified variant: Any variants that meet at least one of the criteria below:
-
(a) A novel variant.
-
(b) A rare variant that has been reported in MITOMAP as a polymorphism, but not in mtDB, or reported in mtDB at a frequency ≤0.2%.
-
(c) A rare variant reported in the literature or MITOMAP as a “mutation” based on a single family study or a single report with no functional studies addressing pathogenicity.
Unclassified variants in this category must be further evaluated by protein structure prediction software (in silico tools) or using other databases ( Figure 2 ):
-
(i) For missense variants, evolutionary conservation and computer-based algorithms are used for in silico prediction of pathogenicity (see Section II below).
-
(ii) If the mtDNA variant occurs in one of the mitochondrial tRNA genes, the Mamit-tRNA database should be used.
Figure 2 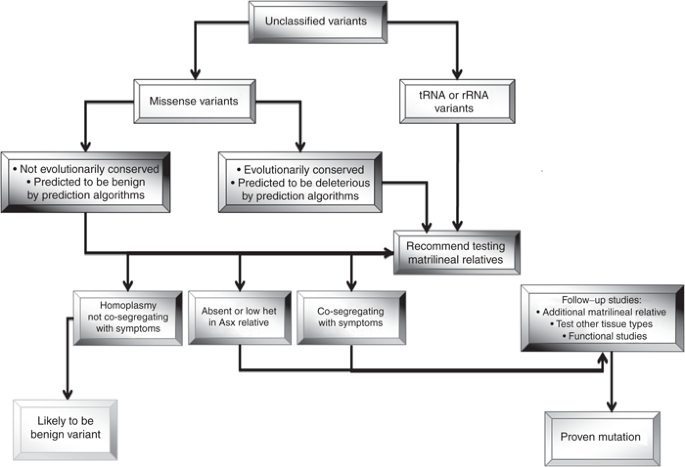
Schematic diagram of follow-up studies for unclassified variants. The unclassified variants should be evaluated by testing matrilineal relatives and additional tissue types and further functional study to access the pathogenicity. ASX, asymptomatic; het, heteroplasmy.
-
-
3. Mutation: mtDNA variants that have been listed in MITOMAP as “confirmed mutations” and have been reported in multiple unrelated patients/families with clinical correlation and/or supporting functional studies.


Classification of mitochondrial DNA variants. mtDNA variants can be classified into three categories, benign variant, unclassified variant, and mutation, based on their frequencies and whether they are reported in databases and the literature. mtDNA, mitochondrial DNA; mtDB, Human Mitochondrial Genome Database.
Interpretation of Missense Variants
Utilization of computational algorhithms, databses, and online resources for the prediction of pathogenicity of missense variants:
-
1
Protein sequence conservation analysis at a specific position is the first step to infer functional importance of an amino acid residue. The protein sequence containing the amino acid of interest should be used as a query for analysis using the NCBI protein blast website (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Select and retrieve orthologous proteins from different species throughout evolution. The different biological species should be selected from phylogenetic trees that represent the similarities and differences in their physical and genetic characteristics. We recommend selecting data for the following species, if available: Homo sapiens, Bos tarurus, Mus musculus/Rattus norvegicus, Gallus gallus, Xenopus laevis, Danio rerio, Drosophila melanogaster, Strongylocentrotus droebachiensis, Caenorhabditis elegans, and Saccharomyces cerevisiae. Use ClustalW2—Multiple Sequence Alignment tool (http://www.ebi.ac.uk/Tools/msa/clustalw2/) for multiple sequence alignment.
-
2
The SIFT algorithm (sorting intolerant from tolerant) (http://sift.jcvi.org) is mainly based on sequence homology. It can be used to predict the likely effect of a nonsynonymous substitution on protein function.11
-
3
The PolyPhen (polymorphism phenotyping) is a structure-sequence-based amino acid substitution prediction method. The current version is PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2). It utilizes the data available in UniProtKB/UniRef100 and is based on conservation, protein folding, and crystal structure.12 This analysis classifies variants as likely benign, possibly damaging, or probably damaging when predicting pathogenicity.
Two caveats exist for the use of predictive software; although limited studies evaluating the reliability of these programs have been reported,13,14 no software has been clinically validated, and the application of software that is based on nuclear-encoded proteins to those that are mtDNA-encoded has the potential for error in that evolutionary conservation of the primary protein sequence is under differing constraints. We evaluated the SIFT and PolyPhen algorithms for reported mtDNA mutations, known benign variants, and unclassified variants ( Table 1 , Figure 3 ). In 21 confirmed mtDNA missense mutations, all 21 (100%) were predicted to be deleterious by SIFT, whereas only 12 (57%) were predicted to be deleterious by PolyPhen. Among the 62 high-frequency nonsynonymous variants, 59 (95%) were predicted to be benign by PolyPhen; however, only 37 (60%) were predicted to be benign by SIFT. The sensitivity and specificity for PolyPhen are 57 and 95% and 100 and 40% for SIFT ( Table 1 ). The concordance rate using PolyPhen and SIFT to predict known mtDNA mutations to be deleterious and known polymorphisms to be benign is ~57% ( Table 2 ).

Use PolyPhen and SIFT to predict mtDNA mutations and variants. The dark shaded areas are proportions of mutations or variants predicted to be deleterious; the light shaded areas are variants predicted to be benign. LHON, Leber hereditary optic neuropathy; mtDNA, mitochondrial DNA; PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant.
Bioinformatics prediction tools may be valuable as screening tools for identifying alleles of high pathogenic potential for molecular and disease association studies. However, because the error rates in both nuclear and mitochondrial predictions are still high, current algorithms do not supplant the need for in vitro or in vivo studies.15
Reporting mtDNA Variants
Use of standard terminology for reporting sequence variants
Reference sequence. Reports should present and describe any detected deleterious mutations and novel/rare variants, provide published reference articles if any, database search results, and in silico predictions to support the variant classification. The clinical report may include all the detected sequence changes that differ from the consensus mitochondrial Cambridge reference sequence (NC_012920). Common benign variants may be tabulated at the end of the report; descriptions and discussions are not necessary.
Nomenclature of mtDNA variants. Standard nomenclature has been significantly revised in recent years. All mutations/variants should be designated according to accepted conventions for the description of sequence variations16 (http://www.hgvs.org/rec.html). Clinical reports should indicate genes involved in each change and describe changes at the nucleotide and protein levels if applicable. Use “m” for mitochondrial DNA nucleotide position and “p” for amino acid position. For example, m.13513G>A (p.D393N, ND5) is a mutation resulting in amino acid substitution of aspartic acid with asparagine at position 393 of the ND5 protein. The m.4418T>C (tRNA Met) represents a variant located at tRNA methionine. Changes at positions that encode overlapping genes or reading frames need to be annotated appropriately.
Heteroplasmy is a unique feature of mitochondrial mutations and variants. In general, the degree of heteroplasmy often corresponds inversely to the pathogenic nature of the variants. Therefore, it is important to indicate whether a change is homoplasmic or heteroplasmic.
Follow-up studies
A suggested follow-up algorithm is presented in Figure 2 .
Test matrilineal relatives. Because of the uncertain biological/clinical significance of unclassified variants, targeted sequence analysis of the patient’s mother and other matrilineal relatives is typically recommended. When a variant is homoplasmic in asymptomatic matrilineal adult relatives and is not cosegregating with disease phenotype, then that variant, by itself, is unlikely to be the primary cause of the clinical symptoms. If a variant is absent or at a low level of heteroplasmy in asymptomatic matrilineal relatives or is cosegregating with disease phenotype, then this variant may be pathogenic. Additional studies, including mitochondrial functional studies and western blot analysis, may be needed to further try to clarify the clinical significance of this variant.
Heteroplasmy quantification and verification. Although the interpretation of a heteroplasmic change is inherently more problematic, if the variant is absent or at lower heteroplasmy in asymptomatic matrilineal relatives, then there is a higher index of suspicion. The degree of heteroplasmy of the variant in various tissues that correlates with the clinical features may also facilitate interpretation. Quantification of the level of heteroplasmy of variant/mutation in tissue samples from invasive (e.g., muscle and skin) or noninvasive (e.g., hair bulbs, urine sediment, and buccal mucosa cells) sources should be considered.
However, it is important to distinguish a low level of heteroplasmy from apparent heteroplasmy because of a technical artifact from poor DNA sequence quality. The same heteroplasmic change should be seen in both sequencing directions by Sanger sequencing and there should be no sequence background (“noise”) in the adjacent regions. If there are multiple heteroplasmic changes detected in one individual, further investigations are needed to verify the heteroplasmic finding: (i) repeat the sequencing using a second DNA extraction to exclude possible contamination of other source of DNA and (ii) contact the referring center to determine whether the patient had a recent blood transfusion. If multiple heteroplasmic changes appear in the same PCR fragment, this could be because of simultaneous amplification of both mtDNA and nuclear homologous regions. The use of alternate PCR primer pairs to resequence the same region is required to avoid the amplification of a nuclear pseudogene region or SNPs at the primer sites that reduce amplification efficiency.
Additional studies. Additional tools and methods can be used to further evaluate the pathogenic potential of mtDNA variants. Electron transport chain complex enzyme assays,17 blue native gel electrophoresis, or western blotting for individual complex protein levels on muscle, liver, or skin fibroblasts can be used to determine (i) whether there are any single or multiple respiratory complex enzyme deficiencies and (ii) whether detected respiratory chain deficiencies correlate with the mtDNA findings (i.e., is there a specific complex deficiency in the presence of a mtDNA-encoded subunit mutation or multiple deficiencies in the presence of a tRNA mutation?). Transmitochondrial cybrid studies and expression studies are also useful in addressing the potential functional impact of a rare or novel variant.
Limitations and notes of sequencing-based mtDNA testing
-
1
A large mtDNA deletion is a common molecular defect of mitochondrial disorders. The deletion sizes range from 3 to 10 kb, with a more common deletion of 5 kb.2,18 Sanger sequencing cannot detect mtDNA large deletions; hence, if a mtDNA deletion is suspected, other methodologies such as Southern blotting, long-range PCR, oligonucleotide array comparative genomic hybridization, or Next-generation sequencing should be considered.
-
2
Sanger sequence may not detect low-level heteroplasmic changes. Conversely, a high level of heteroplasmy may appear as homoplasmy by Sanger sequencing. To precisely determine the degree of heteroplasmy for targeted mutations or variants, other methodologies such as ARMS qPCR,19 PCR-RFLP,20 or Next-generation sequencing should be used.
-
3
When applying Sanger sequencing analysis to the whole mitochondrial DNA, the entire16.6-kb circular mitochondrial genome is usually amplified using 24–30 overlapping amplicons. The primers should be carefully designed and tested to avoid any reported SNPs at the primer sites that may interfere with primer binding. The use of highly diluted template DNA (2–5 ng per 25-µl reaction volume) for PCR is recommended to avoid nonspecific amplification of nuclear homologous sequences.
Based on ACMG recommendations, sequence analysis for a clinical service should be provided by CLIA/CAP-certified laboratories. Interpretation and reporting of sequencing results are limited to qualified providers. We propose a classification scheme ( Figure 1 ) and interpretation guideline based upon variant frequency, in silico predictions, and functional effects.
Disclosure
The authors declare no conflict of interest.
References
Ingman M, Gyllensten U . mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res 2006;34(Database issue):D749–D751.
Ruiz-Pesini E, Lott MT, Procaccio V, et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 2007;35(Database issue):D823–D828.
Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008;10:294–300.
Ingman M, Kaessmann H, Pääbo S, Gyllensten U . Mitochondrial genome variation and the origin of modern humans. Nature 2000;408:708–713.
Ciafaloni E, Ricci E, Shanske S, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 1992;31:391–398.
Brautbar A, Wang J, Abdenur JE, et al. The mitochondrial 13513G>A mutation is associated with Leigh disease phenotypes independent of complex I deficiency in muscle. Mol Genet Metab 2008;94:485–490.
Wilson FH, Hariri A, Farhi A, et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science 2004;306:1190–1194.
Sikorska M, Sandhu JK, Simon DK, et al. Identification of ataxia-associated mtDNA mutations (m.4052T>C and m.9035T>C) and evaluation of their pathogenicity in transmitochondrial cybrids. Muscle Nerve 2009;40:381–394.
Pütz J, Dupuis B, Sissler M, Florentz C . Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 2007;13:1184–1190.
Bandelt HJ, Salas A, Taylor RW, Yao YG . Exaggerated status of “novel” and “pathogenic” mtDNA sequence variants due to inadequate database searches. Hum Mutat 2009;30:191–196.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV . Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res 2006;34:1317–1325.
Tchernitchko D, Goossens M, Wajcman H . In silico prediction of the deleterious effect of a mutation: proceed with caution in clinical genetics. Clin Chem 2004;50:1974–1978.
Hon LS, Zhang Y, Kaminker JS, Zhang Z . Computational prediction of the functional effects of amino acid substitutions in signal peptides using a model-based approach. Hum Mutat 2009;30:99–106.
den Dunnen JT, Antonarakis SE . Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12.
Medja F, Allouche S, Frachon P, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009;9:331–339.
Sadikovic B, Wang J, El-Hattab A, et al. Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes. PLoS ONE 2010;5:e15687.
Wang J, Venegas V, Li F, Wong LJ . Analysis of mitochondrial DNA point mutation heteroplasmy by ARMS quantitative PCR. Curr Protoc Hum Genet 2011;Chapter 19:Unit 19.6.
Mashima Y, Nagano M, Funayama T, et al. Rapid quantification of the heteroplasmy of mutant mitochondrial DNAs in Leber’s hereditary optic neuropathy using the Invader technology. Clin Biochem 2004;37:268–276.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, J., Schmitt, E., Landsverk, M. et al. An integrated approach for classifying mitochondrial DNA variants: one clinical diagnostic laboratory’s experience. Genet Med 14, 620–626 (2012). https://doi.org/10.1038/gim.2012.4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.4
Keywords
This article is cited by
-
Implications of mitochondrial DNA variants in pediatric B-cell acute lymphoblastic leukemia
Egyptian Journal of Medical Human Genetics (2022)
-
Analysis of the entire mitochondrial genome reveals Leber’s hereditary optic neuropathy mitochondrial DNA mutations in an Arab cohort with multiple sclerosis
Scientific Reports (2022)
-
Mitochondrial Genomics: A Complex Field Now Coming of Age
Current Genetic Medicine Reports (2018)
-
Mutations in the two ribosomal RNA genes in mitochondrial DNA among Finnish children with hearing impairment
BMC Medical Genetics (2015)
-
Mitochondrial DNA variant m.15218A > G in Finnish epilepsy patients who have maternal relatives with epilepsy, sensorineural hearing impairment or diabetes mellitus
BMC Medical Genetics (2013)
