
Abstract
Purpose:
Wolfram syndrome is a degenerative, recessive rare disease with an onset in childhood. It is caused by mutations in WFS1 or CISD2 genes. More than 200 different variations in WFS1 have been described in patients with Wolfram syndrome, which complicates the establishment of clear genotype–phenotype correlation. The purpose of this study was to elucidate the role of WFS1 mutations and update the natural history of the disease.
Methods:
This study analyzed clinical and genetic data of 412 patients with Wolfram syndrome published in the last 15 years.
Results:
(i) 15% of published patients do not fulfill the current inclusion criterion; (ii) genotypic prevalence differences may exist among countries; (iii) diabetes mellitus and optic atrophy might not be the first two clinical features in some patients; (iv) mutations are nonuniformly distributed in WFS1; (v) age at onset of diabetes mellitus, hearing defects, and diabetes insipidus may depend on the patient’s genotypic class; and (vi) disease progression rate might depend on genotypic class.
Conclusion:
New genotype–phenotype correlations were established, disease progression rate for the general population and for the genotypic classes has been calculated, and new diagnostic criteria have been proposed. The conclusions raised could be important for patient management and counseling as well as for the development of treatments for Wolfram syndrome.
Genet Med 2013:15(7):497–506
Similar content being viewed by others


Main
Wolfram syndrome (WS; OMIM 222300) is a rare, autosomal recessive, neurodegenerative, and progressive disease.1,2,3 Its prevalence is estimated to be 1 case per 700,000 people in the United Kingdom.4
The minimal ascertainment criterion for diagnosing WS is the coincidence of early onset type 1 diabetes mellitus (DM) (usually during the first decade of life) and bilateral optic atrophy (OA) before the second decade.5 Although these are the two basic symptoms, ~50% patients with WS harbor the full “DIDMOAD” phenotype—the acronym for diabetes insipidus (DI), DM, OA, and deafness (D).6 Because WS is a progressive disease, patients generally develop a wide spectrum of illnesses during their life, such as psychiatric disorders, renal tract abnormalities, nystagmus, ataxia, and loss of deglutition reflex, as well as many others.2,4,7,8,9 Life span is calculated to be 30–40 years,4 and death usually occurs by respiratory failure due to respiratory center atrophy or asphyxia by food aspiration. Several years ago, a particular phenotype was described in some Jordanian families in which patients underwent bleeding gastrointestinal ulceration and lacked DI.8,9
The gene mutated in almost all patients with WS, WFS1, was found and characterized in 1998;10 and mutations in a second gene, CISD2, have been described in those Jordanian patients with gastrointestinal ulceration.11 WFS1 has also been associated with an autosomal dominant form of low-frequency sensorineural hearing loss,12,13,14 autosomal dominant OA,14,15,16 type 2 diabetes,17,18,19 and psychiatric problems.9,15,20
WFS1 has eight exons; the first one is noncoding and comprises 33.4 kbp on chromosome 4p16.1.10 Despite being ubiquitously expressed, differences between tissues with high (i.e., pancreatic β-cells and brain) and with low (i.e., whole blood or kidney) expression are quite significant.10 WFS1 encodes an 890-amino-acid protein called wolframin, a protein with nine transmembrane segments across the endoplasmic reticulum membrane (ER)10 and an Ncytoplasm/CER lumen membrane topology,21,22 which forms homomeric complexes of 400 kDa under physiological conditions.22
Mutations in WFS1 are deleterious for protein expression.23,24 WFS1 depletion in cells induces high levels of endoplasmic reticulum stress, activating the unfolded protein response,25,26 and affects insulin processing and secretion.27,28 Glycosylation sites were identified as important for protein stability,22 and a region that targets unfolded WFS1 to degradation (degron) has been identified.24
The efforts for determining WS natural history have shown the order of onset of the different clinical symptoms.29,30 Nevertheless, the molecular complexity of WS, the wide spectrum of associated diseases, and the small size of patient cohorts (30–60 patients) complicate the establishment of reliable genotype–phenotype correlations. In all attempts, certain WFS1 mutations were associated with milder or with more severe phenotypes.31,32 The data are generally consistent with respect to single cohorts of patients, but some contradictions appear in comparisons of results from different cohorts. Cano et al.33 determined that the presence of two inactivating mutations predispose to an earlier onset of DM and OA. Chaussenot et al.30 showed no association between the effect of the genotype in the neurological signs observed in patients with WS. Rohayem et al.34 showed that the age at onset of DM in patients carrying predicted complete loss of function mutations is lower than those carrying predicted partial or minor loss of function mutations.
In this study, by analyzing and comparing WFS1 mutations and phenotypes of almost 400 published patients with WS, we update the information on the natural history of WS; correlate more clinical features with WFS1 mutations on the basis of a new classification that considers the putative effect of mutations in the protein expression; and estimate, for the first time, the progression rate of the syndrome.
Materials and Methods
Patients
Clinical and genetic data from 412 patients with WS were extracted from 49 references published since 1998 as provided by the authors. All patients were described as having WS. When possible, patients described in more than one publication were identified and their clinical and genetic data were merged into a single record. Only 392 patients were used for the analysis as either age at onset or diagnosis is provided for at least one clinical feature. In all cases, onset and diagnostic age were taken to be equivalent. No information was obtained from patients’ ancestors because it was mainly not mentioned in the publications screened. A list of the references used is provided in Supplementary Table S1 online.
The variations (mutations and polymorphisms) described in the publications have been manually curated, assigned an approved Human Genome Variation Society (HGVS) nomenclature,35 and compared with those listed in the LOVD-WFS1 database.36 We analyzed as mutations (i) c.2020G→A (p.Gly674Arg), considered either a mutation37 or a polymorphism,38 and (ii) c.1367G→A (p.Arg456His), considered either a mutation34 or a polymorphism,31,32,33,39,40 because both are predicted to produce deleterious proteins by Condel41 and PolyPhen-2.42 c.2020G→A is also considered a mutation in the LOVD-WFS1 database.
We considered as polymorphisms c.*47C→T33,43,44 and c.*253G→A37 because they locate to the 3′-untranslated region of WFS1 mRNA, and c.316-37C→T because its effects on splicing have not been indicated.34 Supplementary Table S2 online lists all the mutations described in the patients.
Genotype classification
Mutations were assigned to one of the following types on the basis of their predicted effect on WFS1 expression: (i) type I, which lead to complete depletion of WFS1 protein due to the activation of nonsense-mediated decay,45 including nonsense and frameshift mutations producing stop codons before exon 8 (p.Lys287); (ii) type II, which lead to complete degradation of WFS1 protein by keeping functional the degron in WFS1,24 including all missense mutations except those between amino acids 671–700 and those nonsense mutations producing a stop codon after p.Trp700; and (iii) type III, leading to the expression of a defective or shorter WFS1 protein, including nonsense mutations that introduce a stop codon after exon 8 and before p.Trp700, frameshift mutations introducing a stop codon after exon 8, and missense mutations in amino acids 671–700.
Three main genotypic classes were defined: class A, no WFS1 protein produced; class B, reduced expression of a defective WFS1 protein; and, class C, expression of a defective WFS1 protein. In addition, we subdivided class A into three subclasses: class A1, WFS1 depletion due to WFS1 mRNA degradation; class A2, WFS1 depletion due to mRNA and protein degradation; and class A3, WFS1 depletion due to WFS1 protein degradation. Patients were assigned to a genotypic class on the basis of mutation types as indicated in Supplementary Table S3 online.
Data processing and statistical analysis
All symptoms described in the publications were pooled and categorized into the following clinical features: DM, OA, hearing defects (HD), DI, renal or urological problems (UD), and neurological, psychiatric, or developmental problems (ND). If age was provided for more than one symptom that fit into the same clinical feature, the minimum age was taken as the age at onset.
A life-table approach46 was used for calculating the probability of developing each clinical feature, and the probability of survival was calculated by a Kaplan–Meier approach. Both are available in Supplementary Table S4 online.
The mutation distribution was calculated using a sliding-window strategy. In brief, the amino acids that harbor mutations in a 10-amino-acid window were counted along the protein with a 1-amino-acid step. A total of 881 windows were analyzed and plotted.
Linear regression methods47 were used to calculate the progression rate of the syndrome, in years, as follows: the clinical features of each of the patients, presenting three or more, were ordered by the age at onset and then assigned an ascending number starting at 1. The rate of change in age for each of the patients was the slope of the linear regression between the feature order (x-axis) and the age at onset (y-axis). Median and SEM for each condition were calculated.
Based on the linear regression analysis, the formula for calculating an expected onset age is Agen = slope (n – 1) + Agen=1, where n is the number of clinical features. The onset age of the first feature is taken from the population statistics and corresponds to the median age of the patients within that classification for the first clinical feature.
Data were processed with Microsoft Access and Excel 2007 (Microsoft, Redmond, WA); statistical analysis and some graphical representations were done using R48 or SPSS version 12 (SPSS, Chicago, IL). Random-effects analysis of variance methods were used to assess genotypic class–phenotype correlation. Kruskal–Wallis49 analysis of variance was used to assess differences in ages of patients across genotype–phenotype. Significance level was set at 5%. See Supplementary Figure S4 online for Supplementary Materials and Methods online on western blot.
Results
Wolfram syndrome natural history
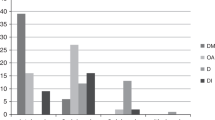
To update the natural history of WS, the clinical data of 412 patients with WS published since 1998 were analyzed. Only patients with WS confirmed in the publications were considered for the analysis. From the 392 patients with age specified for any clinical symptom, 98.21% had DM; 82.14% OA; 48.21% HD; 37.76% DI; 19.39% UD; and 17.09% ND; and 7.40% were deceased at the time of publication (see Supplementary Table S5 online). The population studied presented a median of three clinical features, and 59.44% of the patients had developed 1 to 3 clinical features (see Supplementary Figure S1 online). Onset followed this pattern: DM during the first decade of life; OA during the early second decade; DI and HD, mainly deafness, during the second decade; and UD and ND during the third decade ( Figure 1a and Supplementary Table S5 online), similar to the findings of previous publications.29,30 Although the median age at onset of UD (n = 76) was 20 years, a large proportion of patients developed UD at 10–20 years of age, and three peaks could be observed: one at 13, the second at 21, and the third at 33 years of age. Similarly, ND (n = 67) appeared at 10–30 years of age with a median of 23 years and two peaks—one at 13, and the other at 30 years of age. The median age of deceased patients (DE, n = 29) was 27 ± 11.4 years, but two peaks of higher frequency could be clearly observed—one at 24 and the other at 45 years of age.

Insights into Wolfram syndrome. (a) Natural history of WS. The proportion of patients (density) for each clinical feature at onset age, measured as a nonparametric probability density distribution, is plotted. For descriptive statistics, see Supplementary Table S3 online. (b) Probability of development of WS clinical features by age group. The probability of developing each of the clinical features over age, calculated using a life-table analysis, is displayed. For extended information, see Supplementary Table S4 online. (c) Kaplan–Meier analysis of WS. A Kaplan–Meier approach has been used for analyzing the probability of survival for patients with WS. For extended information, see Supplementary Table S4 online. (d) Order of clinical feature onset in WS. The order in which each clinical feature appears is shown as the percentage of patients with that clinical feature. DM (n = 385); OA (n = 322); HD (n = 189); DI (n = 148); ND (n = 67); UD (n = 72). DE, deceased; DI, diabetes insipidus; DM, diabetes mellitus; HD, hearing defects; ND, neurological, psychiatric, and developmental defects; OA, optic atrophy; UD, urological or renal defects; WS, Wolfram syndrome.
The probability of developing each of the clinical features was then calculated ( Figure 1b and Supplementary Table S4 online). DM, OA, HD, and DI might have onset during the first decade (5% probability at 2–3 years of age for DM, 5–6 for OA, 7–8 for HD, and 8–9 for DI). The probability of developing DM and OA increases sharply during the first two decades (90% probability at 14–15 years of age for DM and at 25–26 years for OA), then reaches a plateau phase (95% probability at 23–24 years of age for DM and at 40–41 years for OA). The risk of developing HD and DI increases steadily until the third decade (at 25–26 years, 60% probability for HD and 50% for DI). From this age on, the probability of developing HD rises, up to 92% at 61–62 years. ND and UD might develop first at the end of the first decade or at the beginning of the second decade (5% probability at 12–13 years of age for UD and 14–15 years for ND). Both probabilities steadily increase up to the fifth decade (at 50 years of age, 53.47% probability of having UD and 64.16% of ND). At 18 years of age, the end of the pediatric age, the probability of having developed the clinical features is: DM, 93.60%; OA, 79.06%; HD, 40.56%; DI, 35.20%; UD, 11.42%; and ND, 7.57%. Life expectancy for patients with WS ( Figure 1c and Supplementary Table S4 online) decreases slowly up to the fifth decade (80.51% at 40 years of age), when a sharp decrease is observed (44.75% at 50 years of age).
The pattern of onset of the clinical features was not followed by all patients ( Figure 1d ). Patients mainly start with DM, although in 20.5%, this was not the first clinical feature. 57.76% mainly developed OA as the second clinical symptom. 41.80% mainly developed HD as the third clinical symptom, although a large number of patients showed it as fourth or fifth clinical manifestation (29.01% and 19.40%, respectively). 35.81% presented DI as the fourth clinical feature, although 33.11% showed it as the third manifestation. UD presented as the fourth or fifth clinical symptom by 27.63% or 28.95%, respectively. ND were detected as the third, fourth, and fifth clinical feature in 20.90, 25.37, and 19.40% of the patients, respectively.
When considering the whole set (392 patients), only 110 (28.06%) showed the full DIDMOAD phenotype. Because the median number of clinical features was three, we considered the possibility that many patients might still not be old enough to develop all four symptoms at the time of publication. Among those patients with four or more clinical features, 66.26% fulfilled the full DIDMOAD phenotype.
We also analyzed whether the current ascertainment criterion, the juvenile onset of DM and OA, was good enough to detect all patients with WS (see Supplementary Figure S2 online). Considering juvenile onset to be onset at ≤18 years of age (the upper limit of pediatric age is 18 years in 15 European Union countries50), only 85.13% (269 of 316) of the patients had both DM and OA.
Genotype distribution
A total of 178 different mutations in WFS1 have been listed in this study in 337 of the 412 patients. This means that 18.20% of patients had either no access to genetic diagnostic tests, no mutations had been identified, or the genetic data were not available at the time of publication. Identification of both mutated alleles was not possible in all cases. Only one mutation was found in WFS1 in 8.31% (28 of 337 patients with described genotype). Four of the mutations present in these patients with WS have also been associated with dominant diseases such as autosomal dominant OA and low-frequency sensorineural hearing loss (see Supplementary Table S6 online).
The 178 mutations are distributed nonuniformly all along the protein, with no major hotspots ( Figure 2a , b ). They concentrate mainly in transmembrane domains, a region important for degradation24 and glycosylation sites. Unexpectedly, some mutations concentrate in two additional regions: (i) at the n-end of the protein comprising amino acids 94–237 and (ii) in the last 100 amino acids ( Figure 2b ), suggesting that these regions might be important for protein function, although deeper and dedicated research would be needed to confirm this point. No founder mutation has been identified, and only six mutations were present in >5% of the patients: c.2649delC (p.Phe884Serfs*68) in 7.42%; c.1230_1233del (p.Val412Serfs*29) in 6.82%; c.409_424dup (p.Val142Glyfs*110) in 6.53%; c.2119G→A (p.Val707Ile) in 6.23%; c.1362_1377del (p.Tyr454*) in 5.64%; and c.1243_1245del (p.Val415del) in 5.34% (see Supplementary Table S2 online).

Mutations distribution in WFS1. (a) Mutation position. Position and type of each mutation is shown in a schematic model of WFS1 protein. The position of the degron (amino acids 671–700),24 the first amino acid encoded by exon 8 (p.Lys287), the two glycosylation sites,22 and the amino acid positions at ER membrane contacts are indicated. Amino acid positions can have more than one mutation and have a different color only if affected by different mutation types. Color code identifies mutation types at that amino acid position: blue, type I mutation; red, type II; green, type III; cyan, type II or III; yellow, type I or II. (b) Mutation distribution. The number of mutations is calculated for a 10-amino-acid sliding window along the protein. White boxes below the chart indicate windows including the transmembrane domains; blue box indicates windows including the degron.24 Glycosylation sites22 are also indicated at their approximated locations. ER, endoplasmic reticulum.
This high heterogeneity complicates setting up a genotype–phenotype correlation for WS. Therefore, we classified these 178 mutations into three mutation types on the basis of the predicted effect they might have on WFS1 protein expression (indicated in the Materials and Methods section). According to this classification, we assigned 5.62% of patient mutations to type I, 55.61% to type II, and 38.20% to type III. A small number of mutations (0.56%) could not be classified into any mutation type because their effect on the protein could not be predicted (see Supplementary Table S2 online).
To study whether different mutation types have a role on the earlier onset of the clinical features, we classified the patients into genotypic classes and subclasses as indicated in the Materials and Methods section. The 337 patients were mainly classified as class A (51.9%), especially class A3, which represents 42.1% ( Figure 3a ). A total of 7.4% were classified as class A1 and 2.4% as class A2. Class B represented 19% and class C, 29.1% of the patients. The distribution of the genotypic classes clearly differed among countries ( Figure 3b and see Supplementary Table S7 online). Patients in France, Germany, and the United Kingdom represented all different genotypic classes, whereas patients in Lebanon, Denmark, and Australia were grouped mainly in subclass A3, patients from Italy mainly in class C, and those from Spain mainly in subclasses A1 and A3. Countries could also be classified into two groups: those in which patients with WS were mostly classified into genotypic classes in which no WFS1 protein is produced (Australia, Denmark, France, Iran, Lebanon, Spain, and the United Kingdom), and those in which a large proportion of patients, >20%, were classified into genotypic class C (Brazil, Germany, Italy, the Netherlands, and Poland).

Genotypic class distribution in patients with Wolfram syndrome. (a) Genotypic class distribution in the whole population. The percentage of patients with each genotypic class and subclass is represented. n = 337. (b) Genotypic class distribution among countries. The number of patients classified into each genotypic class and subclass in some of the countries (those with more than six patients) is presented. For an extension of the data, see Supplementary Table S5 online.
Genotypic class–phenotype correlation
Comparison of the ages at onset for each genotypic class revealed significant differences for DM (P = 0.001) and DI (P = 0.029) when we group the patients into five genotypic classes ( Figure 4 and see Supplementary Table S8 online), and for DI (P = 0.019) and HD (P = 0.038) when grouped them into three genotypic classes (see Supplementary Figure S3a and Supplementary Table S8 online, respectively). A correlation, although not significant, for HD (P = 0.099) when grouped into five genotypic classes and DM (P = 0.072) when grouped into three genotypic classes was also observed. Patients in genotypic class C showed earlier onset of most clinical features; in contrast, patients in genotypic class A1 presented earlier onset of DM, and patients in class A2 developed earlier DI and OA (see Supplementary Table S8 online).

Correlation of phenotype with genotypic classification in Wolfram syndrome. Box-plot representation of patient onset age distribution for each genotypic class and feature. White or black lines in the boxes indicate the median (2nd quartile); box limits show P75 (3rd quartile, top) and P25 (1st quartile, bottom). Outliers (1.5–3× interquartile range) are indicated as circles, and extremes (>3× interquartile range) are indicated as asterisks. Maximum and minimum are indicated by the top and bottom whisker ends, respectively. Number of patients in each category is indicated above x-axis (n). Genotypic classes are A1, A2, A3, B, and C. The probability of different means in each clinical feature, calculated using the Kruskal–Wallis test, is presented. Significant values are in red and values close to significance in bold. For detailed statistics, see Supplementary Table S8 online. DE, deceased; DI, diabetes insipidus; DM, diabetes mellitus; HD, hearing defects; ND, neurological, psychiatric, or developmental defects; OA, optic atrophy; UD, urological or renal defects.
Of note, the genotypic classification of UD and ND, and the deceased patients showed clearly differentiated groups that reflected the bimodal frequencies observed in these clinical symptoms in Figure 1a . In this sense, patients classified as class A2 have an onset of UD at 30 years of age, whereas those classified as class C have onset at 15 years ( Figure 4 ), correlating with the two peaks observed in patient frequency for this clinical feature ( Figure 1a ). Furthermore, patients classified as class A1 have an onset of ND at 30 years of age, and in those in class B or C the onset was at the end of the second decade ( Figure 4 ), which correlates with the two peaks observed in patient frequency ( Figure 1a ). Deceased patients were distributed in two clearly differentiated groups: patients with class A1, with a median age at onset of 40 years, and those with class C, with a median of 25 years of age ( Figure 4 ), similar to the two peaks observed in patient frequency ( Figure 1a ). No differences between sexes were detected (data not shown).
To compare among different genotyping methods, we also correlated genotype and phenotype using other previously described genotypic classifications with this data set (see Supplementary Figure S3b,c and Supplementary Table S8 online). The genotyping methods of Cano et al.33 and Chaussenot et al.30 were based on mutation types and differ from each other only in genotype naming. The genotyping method of Rohayem et al.34 was based on the effect of mutations on the function of WFS1. In the analysis performed, a significant correlation was found only for DM and OA with both methodologies, similar to the findings in the original publications of Cano et al.33 and Chaussenot et al.30 and expanding the findings in Rohayem et al.34 By contrast, a significant correlation for DI and HD (see Supplementary Figures S3a and Supplementary Table S8 online) was observed when grouped into three genotypic classes and for DM and DI when grouped into five genotypic classes ( Figure 4 and see Supplementary Table S8 online) with the approach described here.
Clinical progression of Wolfram syndrome
Although there is indisputable interest in knowing whether the evolution of the disease depends on the different genetic backgrounds, the evolution rate of patients with WS through the different clinical features is still unknown. We therefore used a regression approach, as indicated in the Materials and Methods section, on those patients harboring three or more clinical features, as a way to estimate the evolution rate of WS in the general population ( Figure 5 , shadowed line). We obtained a progression rate of 3.90 ± 0.222 (n = 260) for the whole set of patients analyzed, indicating how fast patients with WS develop the clinical features. We then estimated the rate for the different genotypic classes. If the estimated rate is smaller than that of the general population, it would mean that the onset of the different clinical features is earlier, and, therefore, the overall phenotype might be worse as more manifestations of the disease have started. When the genotypic classification previously defined was considered for the estimation of the rate ( Figure 5 and Supplementary Table S9 online), the progression rate of the disease through the clinical features was significantly faster in patients in genotypic class C (3.50 ± 0.261, n = 69), and faster for classes A2 (2.90 ± 1.443, n = 7) and B (3.60 ± 0.592, n = 48) than the general population. Those in genotypic class A1 (4.90 ± 0.796, n = 21) showed a slower progression rate than the general population of patients with WS and patients in genotypic class A3 (4.0 ± 0.376, n = 112).

Progression rate of Wolfram syndrome. The progression rate of patients with Wolfram syndrome with three or more clinical features sorted by the age of onset is calculated as the slope of the linear regression that binds them. Median ± SEM is shown for each genotypic class inside the histograms. The slope was calculated from 21 patients in class A1; 7 in class A2; 133 in class A3; 49 in class B; and 70 in class C. The median slope ± SEM for all the patients (n = 260) is shown as a dashed line and gray area, respectively. Slopes that are higher than the population median show slower progression rates than the general population, and those that are smaller show faster progression rates. *P = 0.009.
Discussion
The data published over the past 15 years allowed us to carry out a deep study on the onset of the clinical features and to describe how patients with WS evolved thanks to the large number of patients described. The approach used has its pitfalls due to differences between publications in (i) the amount of data reported, (ii) the age when the different clinical analysis are done, (iii) the clinical threshold for presenting the different symptoms, and (iv) the accuracy of the onset ages of some clinical features due to unrecognized subtle changes (i.e., ND) or different clinical thresholds (i.e., UD), among other reasons. Getting a well-characterized cohort that is managed under standardized criteria and large enough to allow the analysis presented here with high-quality data will not be possible until further population and integration of the disease-associated registers recently initiated in the United States, Europe, and Spain, due to the low prevalence of WS.
The picture of the natural history of the disease obtained here is in agreement with the ones previously published29,30 and extend the knowledge. The main differences observed are in the onset of UD and ND, although detailed age of onset was only provided for a limited number of patients (76 of 148 with UD and 67 of 141 with ND), complicating an accurate analysis. In the set of patients used here, the median age at onset for UD was 20 years (n = 76), similar to Barrett’s series (n = 30) and different from the 12 years in Chaussenot’s series (n = 59). The peaks detected on the frequency of patients with UD ( Figure 1a ) correlated with the median onset age in genotypic class A1 or C for the first (13 years), class B for the second (21 years), and A2 for the third peak (33 years, Figure 4 ).
Similarly, the onset of ND also differs with previous data. In Chaussenot’s series, the onset was 15 years of age (n = 30),30 half the age of Barrett’s series (n = 12).29 In the set of patients used here the two peaks detected, one at 13 and the other at 30 years of age ( Figure 1a ), correlated with the median age of class A1 for the first peak and class B or C for the second one ( Figure 4 ). These results together suggest that different genotypic classes could be behind these differences in the frequency distribution of the clinical features. More detailed phenotype information is therefore needed for refining these observations.
Among all patients analyzed, 66.26% had the full DIDMOAD phenotype when considering only those with four or more symptoms. This number is higher than the one previously reported,6 and indicates that one-third of patients with WS might not show the four classical clinical features defining the syndrome.
The current ascertainment criterion for WS is the juvenile onset of DM and OA. Considering juvenile onset as equivalent to age 18,50 almost 15% of the patients in the present series who had both clinical features did not fulfill this criterion (see Supplementary Figure S2 online), indicating that people affected by this disease might be underestimated. Furthermore, the calculated probability of developing each of the clinical features at 18 years of age for DM and OA is not 100% ( Figure 1b and Supplementary Table S4 online). We suggest changing this criterion. To this end, we foresee two possibilities: (i) to consider patients with DM and OA at any age. In this situation, 96.05% of the patients would be included (316 of 329 patients with two or more clinical features); or (ii) to take patients with two of any of the DIDMOAD symptoms. Then, 98.78% of the patients would be included (325 of 329 patients with two or more clinical features). We propose the latter criterion to be used for considering WS because it may help to genetically diagnose almost all patients with WS earlier and might allow detection of atypical WS patients.15,16,51 Furthermore, 20% of patients with WS do not debut with DM but other clinical features, and a large proportion do not develop as a second clinical feature DM or OA ( Figure 1d ). All these data together suggest a wider heterogeneity in WS than previously demonstrated by other studies and has important implications in the clinical specialties that have to be aware of this disease.
Based on predicting the effect of WFS1 mutations on protein expression using current knowledge on cell functioning and in WFS1 protein stability, we have classified the mutations into three different types. Type I mutations are those that activate the mechanism used by the cell to degrade aberrant mRNAs (nonsense-mediated decay), leading to the absence of wolframin due to mRNA degradation. Although it has not been demonstrated in patients with WS, those mutations that lead to the creation of a premature stop codon before the beginning of the last exon (exon 8 in WFS1) are at risk for activating this mechanism.45,52
We have classified remaining mutations into type II or III, based on the recent finding of a sequence that targets unfolded WFS1 to degradation (degron) between amino acids 671 and 700.24 Differences in protein stability have already been demonstrated for some previously published WFS1 mutations. Heterologous expression in human cells of WFS1 mutants that keep this degron (type II mutations), such as p.Pro504Leu, p.Arg629Trp, p.Trp700*, p.Pro704Leu, p.Pro724Leu, p.Glu737Lys, p.Phe883*, and p.Pro885Leu, led to cellular depletion or reduced expression of WFS1 protein.23,24 By contrast, the expression of mutants that affect this region (type III mutants), such as p.Trp648*, p.Tyr660*, and p.Gln667*, was similar to the expression of the wild-type protein.23,24
Furthermore, WFS1 expression in previously published patients supports the mutation and genotypic classifications we propose. In this sense, a patient predicted to be classified into genotypic class B for carrying heterozygous type II and type III mutations (p.Arg629Trp and p.Trp371*, respectively) showed a reduced content of a defective WFS1 protein. Furthermore, a patient classified genotypically into class A3 (homozygous for p.Trp700*, a type II mutation) showed no WFS1 protein expressed,23 in concordance also with the heterologous expression of this mutant. In addition, as predicted, a patient homozygous for a type II mutation, c.2452C→T (p.Arg818Cys), and classified into class A3, showed no WFS1 protein expressed (see Supplementary Figure S4 online).
The lack of one mutant allele in 8.31% of the patients points to: (i) the lack of detection of a second mutation, (ii) the existence of dominant mutations, or (iii) mutations in another gene. Because no data are available on any multiplex ligation–dependant probe amplification analysis performed on these patients, the presence of deleted WFS1 regions is possible. In addition, the effect on WFS1 expression of those variations located in the promoter or 5′- or 3′-untranslated regions of WFS1 mRNA and the effect on WFS1 localization and activity of many variations are still unknown. We could then envision that new classes and subclasses based on WFS1 protein localization, activity, or expression might need to be defined. A functional assay allowing further and intensive research in the effect of WFS1 variations is needed for properly classifying all variations.
The genotypic class–phenotype correlation performed indicates that the differences observed in the age at onset of DM, DI, and HD correlates with the genotypic classification performed. We could not exclude a role in the other clinical features analyzed (UD, ND, and deceased); additional patients with these features should be evaluated. For comparison purposes, we also performed the genotype–phenotype correlation using other described genotypic classifications30,33,34 with the same data set. In the analysis performed, a significant correlation was found only for DM and OA with all methodologies tested, similar to the findings in the original publications of Cano et al.33 and Chaussenot et al.30 and expanding the findings of Rohayem et al.34 By contrast, we observed significant correlations in DM, DI, and HD with our genotyping strategy ( Figure 3 and Supplementary Figure S4 and Supplementary Table S8 online). All this suggests that the depletion or expression of a mutated form of WFS1 might play a role in determining the age at onset of these clinical features in patients with WS.
It was possible, for the first time, to estimate the rate at which the patients developed the different clinical features in WS. Of note, this progression rate varied in accordance with the patient’s genotypic class, providing the first clues that might explain the differences in the development of the phenotype between patients. Furthermore, the calculation of age at onset of the clinical manifestations based on this estimation and the genotypic classification gets very close to the observed median for that genotype (see Supplementary Table S9 online). These data support our genotyping approach and progression-rate estimation, although the accuracy and predictive value of the progression rate would be highly improved if the amount of data and the comparative quality of data were increased.
Because WS is a rare disease, it is difficult to study patient cohorts that are large enough without the cooperation of researchers from different countries. This means that normalized, detailed, and high-quality phenotypic and genotypic information should be recorded for all patients. The initiatives from the European53 and Spanish registers (EURO-WABB (http://www.euro-wabb.org) and REWBA (https://rewba.idibell.cat), respectively) could provide, in the near future, the data to address these and other unsolved questions. Definitive knowledge about the natural history of WS will come from the integration of the data in the aforementioned registers and those from other countries, such as the one in the United States (https://wolframsyndrome.dom.wustl.edu/medical-research/Wolfram-Syndrome-Home.aspx), into an international register. The results presented in here, although based on questionable quality of data, provide the deepest view of the natural history of WS obtained with currently available data.
WS is a monogenic disease, and its characteristics could make it a useful model for understanding more complex diseases such as DM, OA, HD, and other neurologically related clinical symptoms. The described genotypic classification of patients helps in understanding the observed differences between patients.
Disclosure
The authors declare no conflict of interest.
References
Page MM, Asmal AC, Edwards CR . Recessive inheritance of diabetes: the syndrome of diabetes insipidus, diabetes mellitus, optic atrophy and deafness. Q J Med 1976;45:505–520.
Cremers CW, Wijdeveld PG, Pinckers AJ . Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl 1977;264:1–16.
Fraser FC, Gunn T . Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome? J Med Genet 1977;14:190–193.
Barrett TG, Bundey SE, Macleod AF . Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995;346:1458–1463.
Wolfram D, Wagener H . Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clinic Proc. 1938;13:715–718.
Pilley SF, Thompson HS . Familial syndrome of diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (didmoad) in childhood. Br J Ophthalmol 1976;60:294–298.
Swift RG, Sadler DB, Swift M . Psychiatric findings in Wolfram syndrome homozygotes. Lancet 1990;336:667–669.
El-Shanti H, Lidral AC, Jarrah N, Druhan L, Ajlouni K . Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am J Hum Genet 2000;66:1229–1236.
Khanim F, Kirk J, Latif F, Barrett TG . WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat 2001;17:357–367.
Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998;20:143–148.
Amr S, Heisey C, Zhang M, et al. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 2007;81:673–683.
Bespalova IN, Van Camp G, Bom SJ, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001;10:2501–2508.
Young TL, Ives E, Lynch E, et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001;10:2509–2514.
Hogewind BFT, Pennings RJE, Hol Fa, et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Molecular Vision 2010;16:26–35.
Rendtorff ND, Lodahl M, Boulahbel H, et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A 2011;155A:1298–1313.
Eiberg H, Hansen L, Kjer B, et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet 2006;43:435–440.
Wasson J, Permutt MA . Candidate gene studies reveal that the WFS1 gene joins the expanding list of novel type 2 diabetes genes. Diabetologia 2008;51:391–393.
Lauenborg J, Grarup N, Damm P, et al. Common type 2 diabetes risk gene variants associate with gestational diabetes. J Clin Endocrinol Metab 2009;94:145–150.
Chistiakov DA, Khodyrev DS, Smetanina SA, Bel’chikova LN, Suplotova LA, Nosikov VV . A WFS1 haplotype consisting of the minor alleles of rs752854, rs10010131, and rs734312 shows a protective role against type 2 diabetes in Russian patients. Rev Diabet Stud 2010;7:285–292.
Middle F, Jones I, McCandless F, et al. Bipolar disorder and variation at a common polymorphism (A1832G) within exon 8 of the Wolfram gene. Am J Med Genet 2000;96:154–157.
Takeda K, Inoue H, Tanizawa Y, et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10:477–484.
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF . Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 2003;12:2003–2012.
Hofmann S, Bauer MF . Wolfram syndrome-associated mutations lead to instability and proteasomal degradation of wolframin. FEBS Lett 2006;580:4000–4004.
Guo X, Shen S, Song S, et al. The E3 ligase Smurf1 regulates Wolfram syndrome protein stability at the endoplasmic reticulum. J Biol Chem 2011;286:18037–18047.
Fonseca SG, Fukuma M, Lipson KL, et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 2005;280:39609–39615.
Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 2010;120:744–755.
Ishihara H, Takeda S, Tamura A, et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 2004;13:1159–1170.
Hatanaka M, Tanabe K, Yanai A, et al. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet 2011;20:1274–1284.
Barrett TG, Bundey SE . Wolfram (DIDMOAD) syndrome. J Med Genet 1997;34:838–841.
Chaussenot A, Bannwarth S, Rouzier C, et al. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol 2011;69:501–508.
Smith CJ, Crock PA, King BR, Meldrum CJ, Scott RJ . Phenotype-genotype correlations in a series of Wolfram syndrome families. Diabetes Care 2004;27:2003–2009.
Giuliano F, Bannwarth S, Monnot S, et al.; French Group of WS. Wolfram syndrome in French population: characterization of novel mutations and polymorphisms in the WFS1 gene. Hum Mutat 2005;25:99–100.
Cano A, Rouzier C, Monnot S, et al.; French Group of Wolfram Syndrome. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A 2007;143A:1605–1612.
Rohayem J, Ehlers C, Wiedemann B, et al.; Wolfram Syndrome Diabetes Writing Group. Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes Care 2011;34:1503–1510.
den Dunnen JT, Antonarakis SE . Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12.
LOVD-WFS1 database. https://lovd.euro-wabb.org/home.php?select_db=WFS1. Accessed 8th November 2012.
Aloi C, Salina A, Pasquali L, et al. Wolfram syndrome: new mutations, different phenotype. PLoS ONE 2012;7:e29150.
Gómez-Zaera M, Strom TM, Rodríguez B, Estivill X, Meitinger T, Nunes V . Presence of a major WFS1 mutation in Spanish Wolfram syndrome pedigrees. Mol Genet Metab 2001;72:72–81.
Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998;20:143–148.
Hardy C, Khanim F, Torres R, et al. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 1999;65:1279–1290.
González-Pérez A, López-Bigas N . Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 2011;88:440–449.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Hansen L, Eiberg H, Barrett T, et al. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet 2005;13:1275–1284.
Ganie MA, Laway BA, Nisar S, et al. Presentation and clinical course of Wolfram (DIDMOAD) syndrome from North India. Diabet Med 2011;28:1337–1342.
Brogna S, Wen J . Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 2009;16:107–113.
Klein J, Moeschberger M . Survival Analysis: Techniques for Censored and Truncated Data. Springer-Verlag: New York, 1997.
Weisberg S . Applied Linear Regression. 3rd edition ed. John Wiley & Sons, Inc.: New York, 2005.
R project. http://cran/r-project.org.
Kruskal WH, Wallis WA . Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583.
van Esso D, del Torso S, Hadjipanayis A, et al.; Primary-Secondary Working Group (PSWG) of European Academy of Paediatrics (EAP). Paediatric primary care in Europe: variation between countries. Arch Dis Child 2010;95:791–795.
Tranebjaerg L, Barrett T, Rendtorff ND . WFS1-Related Disorders, 1993.
Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE . NMD: RNA biology meets human genetic medicine. Biochem J 2010;430:365–377.
Maffei P, Farmer A, Milan G, et al. The EURO-WABB project. Paper presented at: 15th International & 14th European Congress of Endocrinology (ICE/ECE 2012), 2012; Florence, Italy.
Acknowledgements
We thank Tim Barrett, Clara Vilches, and Raquel Guillén for critical reading of the manuscript; Selei Gharanei and Malgosia Zatyka for WFS1 blotting conditions; and Isidro Ferrer and the Biobanco IDIBELL for the control brain sample. We also thank the family of the WS patient for donating the brain sample and Nolwenn Jaffré for encouragement and motivation. This work was supported by SAF2009-12606-C02-02 from MINECO (Spain), INTRA/10/730.1 from CIBER de Enfermedades Raras (an initiative of ISCIII, Spain), and SGR 2009-1490 (Generalitat de Catalunya). Partial support was given by the EURO-WABB project, which has received funding from the European Union, in the framework of the Health Programme.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Table 1
(PDF 51 kb)
Supplementary Table 2
(PDF 86 kb)
Supplementary Table 3
(PDF 18 kb)
Supplementary Table 4
(XLS 75 kb)
Supplementary Table 5
(PDF 22 kb)
Supplementary Table 6
(PDF 43 kb)
Supplementary Table 7
(PDF 24 kb)
Supplementary Table 8
(PDF 58 kb)
Supplementary Table 9
(PDF 31 kb)
Supplementary Figure 1.
(PDF 346 kb)
Supplementary Figure 2.
(PDF 411 kb)
Supplementary Figure 3.
(PDF 234 kb)
Supplementary Figure 4.
(PDF 174 kb)
Rights and permissions
About this article
Cite this article
de Heredia, M., Clèries, R. & Nunes, V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 15, 497–506 (2013). https://doi.org/10.1038/gim.2012.180
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.180
Keywords
This article is cited by
-
Wfs1E864K knock-in mice illuminate the fundamental role of Wfs1 in endocochlear potential production
Cell Death & Disease (2023)
-
The genetic and clinical characteristics of WFS1 related diabetes in Chinese early onset type 2 diabetes
Scientific Reports (2023)
-
Unique three-site compound heterozygous mutation in the WFS1 gene in Wolfram syndrome
BMC Endocrine Disorders (2021)
-
A mutant wfs1 zebrafish model of Wolfram syndrome manifesting visual dysfunction and developmental delay
Scientific Reports (2021)
-
Wolfram syndrome 1 in the Italian population: genotype–phenotype correlations
Pediatric Research (2020)
