
Abstract
Adoption of whole-genome sequencing as a routine biomedical tool is dependent not only on the availability of new high-throughput sequencing technologies, but also on the concomitant development of methods and tools for data collection, analysis, and interpretation. It would also be enormously facilitated by the development of decision support systems for clinicians and consideration of how such information can best be incorporated into care pathways. Here we present an overview of the data analysis and interpretation pipeline, the wider informatics needs, and some of the relevant ethical and legal issues.
Genet Med 2013:15(3):165–171
Similar content being viewed by others
Introduction
Technological advances have resulted in a dramatic fall in the cost of human genome sequencing. However, the sequencing assay is only the beginning of the process of converting a sample of DNA into meaningful genetic information. The next step of data collection and analysis involves extensive use of various computational methods for converting raw data into sequence information, and the application of bioinformatics techniques for the interpretation of that sequence. The enormous amount of data generated by massively parallel next-generation sequencing (NGS) technologies has shifted the workload away from upstream sample preparation and toward downstream analysis processes. This means that, along with the development of sequencing technologies, concurrent development of appropriate informatics solutions is urgently needed to make clinical interpretation of individual genomic variation a realistic goal.1,2
The Data Analysis Pipeline
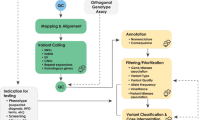
The data analysis process can be broadly divided into the following three stages ( Figure 1 ).

Outline of informatics pipeline for processing and analyzing data from massively parallel sequencing platforms.
Primary analysis: base calling
That is, converting raw data, based on changes in light intensity or electrical current, into short sequences of nucleotides.3
Secondary analysis: alignment and variant calling
That is, mapping individual short sequences of nucleotides, or reads, to a reference sequence and determining variation from that reference.4
Tertiary analysis: interpretation
That is, analyzing variants to assess their origin, uniqueness, and functional impact.5
Each of these steps requires purpose-built databases, algorithms, software, and expertise to perform. By and large, issues related to primary analysis have been solved and are becoming increasingly automated, and are therefore not discussed further here. Secondary analysis is also becoming increasingly automated for human genome resequencing, and methods of mapping reads to the most recent human genome reference sequence (GRCh37), and calling variants from it, are becoming standardized. The major bottleneck for wider clinical application of NGS is the interpretation of sequence data, which is still a nascent field in terms of developing algorithms, appropriate analytical tools and effective evidence bases of human genotype–phenotype associations. Although the steps involved in interpretation and application of results will vary depending on the specific clinical setting, the purpose of the testing, and the clinical question, it is likely that there will be commonalities in both the basic analysis pipeline and tools used. Similarly, although some of the details may change with the introduction of third-generation sequencing technologies, such as those that involve real-time detection of single molecules, the data analysis challenge posed by massively parallel sequencing technologies will remain essentially the same. Below, we provide an overview of the secondary and tertiary steps involved in analyzing raw sequence reads from NGS technologies.
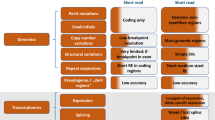
Secondary Analysis: Variant Calling and Annotation
Following initial base calling, the next step toward generating useful genetic information from sequencing reads (i.e., short sequences of nucleotides) involves assembly of the reads into a complete genome sequence by comparison of multiple overlapping reads and the reference sequence. Longer read lengths would make this process much simpler, as each read would be much more likely to map uniquely to a single position in the genome. The process is complicated by the extent of variation in the sample sequence from the reference sequence, and generating a list of true variants is made challenging because there are several possible explanations for differences between the reference and sample genome:
-
Inaccuracies in the reference genome, which does not represent any single individual and is still incomplete due to highly repetitive regions that have yet to be sequenced. Furthermore, to date, the reference sequence has been regularly updated, which has caused specific regions to map to different locations between versions.
-
Incorrect base calls in the sample sequence due to sequencing or amplification errors.6,7,8 However, this can be mitigated through read-depth analysis (i.e., evaluating the number of times an individual base is sequenced in independent reads) to assess the reliability of each base call.
-
Incorrect alignment between the reference and sample may arise due to the highly repetitive nature of substantial portions of the genome and (in the case of NGS platforms) short read lengths. This can result in individual reads mapping to multiple locations. The likelihood that a read is mapped correctly can be indicated by a mapping score, and the mapping reliability can be substantially improved by using paired-end reads (i.e., sequencing two ends of the same DNA molecule) to assess the presence of insertions and deletions.8,9
-
Variation may represent true genetic variation in the sample. As each human genome is estimated to differ from the reference sequence in ~3–4 million sites,9,10 including single-nucleotide changes and structural variants, mapping these variants is a challenge. Determining which variants are derived from the same physical chromosome can also be difficult from short reads, and haplotypes must either be assembled11 or imputed.12 Using paired-end reads for mapping is particularly important for identifying structural variation, and is critical for cancer genome sequencing due to the presence of extensive large structural rearrangements relative to the matched germline genome, including both intra- and interchromosomal rearrangements.13
The addition of biological information to sequence data is an important step toward making it possible to interpret the potential effects of variants, and involves a mixture of automatic annotation by computational prediction and manual annotation (curation) by expert interpretation of experimental data. The main steps relate to structural information (e.g., gene location, structure, coding, splice sites, and regulatory motifs) and functional information (e.g., protein structure, function, interactions, and expression).14 A fully annotated sequence can include an enormous amount of information, including common and rare variants, comparisons with other species,15 known genetic and epigenetic variants, regulatory features, transcript, and expression data, as well as links to protein databases. However, annotation is currently both incomplete and imperfect in the human genome. Projects such as ENCODE, the Encyclopedia of DNA Elements,16,17 and the related GENCODE, encyclopedia of genes and gene variants,18,19 aim to identify all functional elements in the human genome sequence and will ultimately be used to annotate all evidence-based gene features in the entire human genome at a high accuracy.
Numerous programs have been developed specifically for genome assembly, alignment, and variant calling based on DNA sequence reads from high-throughput next-generation sequencing platforms2,4,20,21 ( Table 1 ). These include software developed for use with a particular sequencing platform, open access academic software with a variety of functionalities and platform compatibilities, and proprietary software designed for specific purposes such as diagnostics ( Table 2 ). A major issue for standard alignment programs is the interpretation of small insertions and deletions, which has been partly addressed by the development of new programs specifically for this purpose22; however, current technology does not yet allow for confident analysis of interpretation of small insertions and deletions as long as several hundred base pairs, especially repeat sequences. Various dedicated software packages have also been developed specifically for cancer genome assembly and variant calling, which take into account factors such as genetic heterogeneity within the sample.23
Sequence information can be visualized through a graphical interface or genome browser such as Ensembl24 and the UCSC Genome browser,25 which are extensively used for research purposes and provide the most recently assembled build of the human genome, with information on gene location, intron/exon boundaries, data on RNA expression, common single-nucleotide polymorphisms and copy number variants, mutations, and alignments with other species. They are extensively hyperlinked to external databases, are actively curated and regularly updated, and essentially act as portals for accessing and exploring annotated reference genomes and databases. Numerous companies have developed proprietary genome browsers that enable a sample genome to be viewed, annotated, and compared directly against the reference genome; some proprietary browsers accompany particular sequencing platforms, others have been developed for specific markets (such as the medical diagnostics industry), and some are freely available and can be adapted by the user to fit various purposes.
Tertiary Analysis: Interpreting the Data in the Context of an Individual
Interpreting genomic data involves analyzing variants to assess their origin, uniqueness, and likely functional impact. This is aided by tools such as databases of genomic variation (both normal and pathogenic), algorithms for evaluating likely pathogenicity of particular mutations and tools such as the Variant Effect Predictor (VEP) available via Ensembl. Genomic analysis for clinical purposes usually attempts to identify likely pathogenic mutation(s) that account for a specific phenotype. Although clinical interpretation of whole-genome sequence data is still in its infancy, clinical interpretation of very large structural variants identified through karyotyping or DNA microarrays is now standard practice.26 Structural variants uncovered through these older genome-wide technologies are a priori more likely to be pathogenic due to the relatively low resolution of these technologies and the limited number of large structural variants in the normal population.27 Clinical interpretation of rare structural variation is already substantially aided by databases such as DECIPHER,28 which allows the phenotypic consequences of overlapping duplications and deletions in different patients from around the world to be compared.
In contrast, determining the most likely causal variant(s) among a plethora of sequence-level variants of unknown clinical significance—which include both normal and pathogenic variation—is extremely difficult. The first step in such an analysis is to filter out known (or suspected) nonpathogenic variation. Initially, genetic and functional filters can be applied to exclude common, nonpathogenic, or irrelevant variants and those that are not expected to have a functional effect.5,29
Genetic filter
Comparison with databases of genomic variation30 ( Table 3 ), unrelated individuals with the same phenotype, the individual’s germline genome in the case of somatic sequencing, and additional analysis of family members to determine inheritance can lead to the identification of candidate variants.
Functional filter
Analysis of the genomic or exonic location of the variant, evolutionary conservation, and predicted effect on protein structure, function, or interactions allows the exclusion of variants that are expected to have no known functional effect. This could include: evaluation of evolutionary conservation31; prediction of the effect of splice site disruptions; prediction of haploinsufficiency status of genes32; investigation of the expression of RNA or protein in the relevant tissue; use of functional models to investigate the phenotypic effect of gene knockouts; assessment of the role of the protein in relevant biochemical networks and pathways; and prediction of the effect of amino acid substitutions caused by nonsynonymous changes on protein stability, structure, and function based on physical and comparative methods.33,34,35
Various sequence analysis platforms have been developed that integrate and automate many of these processes, including those for use in medical diagnostics. However, most will result in numerous candidate variants, and a final interpretation by a clinician and/or clinical scientist must integrate biological knowledge with relevant phenotypic and clinical information to assess the relevance of any candidate variant(s) to decisions regarding appropriate interventions. This might include the inheritance, heritability, penetrance, and expressivity of the variant, as well as implications for therapeutic options and treatment regimes.
Decision Support for Clinicians
Most existing informatics and database resources have been developed for research purposes and are used in limited clinical settings such as specialist clinical genetics services. Implementing NGS technologies and whole-genome sequencing for routine diagnostics requires a stable, clinical-grade sample tracking and analysis pipeline, equivalent to the laboratory accreditation system, to ensure reliable performance and accuracy. Key to this is the existence of extensive databases of both normal and pathogenic variation, to allow partially automated interpretation of individual variants.
Ensuring interoperability between the plethora of sequencing platforms, databases, and analysis tools presents a major hurdle that must be overcome. This is partly being addressed by the development of standardized ontologies by organizations such as the Human Genome Organization nomenclature committee,36 the Human Genome Variation Society, the US National Center for Biomedical Ontology, the Genome Ontology consortium,37 and the Human Phenotype Ontology project.38 In addition, initiatives such as the international Human Variome Project,39 ELIXIR at the European Bioinformatics Institute, and the EU-FP7 Gen2Phen project are working on models and standards in data description, storage, and integration for life science and biomedical databases, although, in general such attempts at standardization are still lacking in cancer genomics. However, the International Society for Gastrointestinal Hereditary Tumours Incorporated (InSiGHT) and Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) are aiming to address this gap with regard to gastrointestinal tumors and breast cancer, respectively.
The routine use of genomic information in a clinical setting also requires integration with other initiatives such as the Unified Medical Language System, Systematized Nomenclature of Medicine—Clinical Terms (SNOMED CT), Logical Observation Identifiers Names and Codes (LOINC), and Health Level 7 initiatives, which have been integral in the development of a common language for electronic health records to allow the appropriate retention, integration, processing, and exchange of unambiguous medical data.
Together with the creation of interoperable systems, it is likely that widespread use of genomics in the clinical setting will require appropriate decision support systems to help clinicians interpret plausibly pathogenic genomic variants, integrate genomic information into the patient pathway, and guide preventative and therapeutic options, both for diagnosis and personalized/stratified treatments. Most clinical decision support systems consist of three parts: a dynamic knowledge base, an inference engine based on an agreed set of rules, and an appropriate mechanism for communication with the health-care professional (or patient).40 In genomic terms, this might equate to: a database (or databases) of genotype–phenotype associations, an analysis pipeline to prioritize a list of candidate variants of interest in a particular patient, and a user-friendly portal for inputting, accessing, and visualizing patient data. Standardized representation of genomic and nongenomic patient data is essential to ensure reliable computer-based interpretation and processing,41 and robust epidemiological data and statistical methods are required to ensure evidence-based analysis.
Ultimately, the value of any clinical decision support system is heavily dependent on the robustness of the knowledge base, which must be regularly updated and maintained. Given the enormous number of variants in every genome, most of which are common, most variants (including most private mutations) are likely to be benign. Genetic variants are likely to fall into three broad categories: those with a clear clinical interpretation (mostly relating to well-characterized variants associated with monogenic disorders), those plausibly associated with disease but with unknown or insufficiently proven clinical significance, and those with no known association with disease. There will be regular movement of variants between these categories as new discoveries are made and genotype–phenotype associations cataloged. Although attempts are being made to develop standardized categorization, variant repositories, and evidence bases as described above, there is currently no standardized process or system for assigning and annotating this categorization, no centralized curated repository for genes or variants associated with specific diseases, and frequently a lack of data on which to make an evidence-based assessment of the clinical validity and utility of any individual test or analysis. Thus, current practice remains heavily dependent on the knowledge and skill of individual practitioners.
Data Sharing and Privacy
The widespread application of sequencing in a clinical setting is dependent on robust, extensive, and transparent databases of population genome variation and genotype–phenotype correlations containing anonymized information. However, many applications also depend on the storage of individual linked genomic data of relevance to diagnosis, prognosis, and management of disease(s) in an individual. In practice, harnessing genomic data for health benefit is likely to involve data sharing to different extents between multiple parties (including laboratory staff, informaticians, researchers, clinicians, patients, and their family members) across multiple jurisdictions. This complexity reflects fluidity of the boundaries between research and clinical implementation, as technologies are developed, and genetic variants are analyzed, annotated, interpreted, and validated for clinical use.
Concerns about the use of automated pipelines to facilitate data management, processing, and interpretation arise partly as a consequence of the distinctive characteristics of genomic data. These include the potential identifiability of genomic (and often associated phenotypic) data, the immutability of the data throughout an individual’s lifetime, the potential predictive capability of the data, and the wider possible impact of the data on the family of the individual undergoing testing.42 These features raise questions about the proportionate safeguards and governance that should be put in place to limit data access and security while respecting patient privacy and confidentiality.42 These issues are not unique to genomic data, but apply equally to predictive health data of all types.43
Nevertheless, the degree of protection that should be afforded to genomic data is a continuing challenge, particularly because data protection legislation within Europe places limits on processing data that are personally identifiable but allows more liberal access to data that are anonymous. Thus the extent to which genomic data, including whole-genome sequence data, are capable of being anonymized has profound implications because these technologies are translated into clinical settings. Methods applied for de-identification in a research context, such as the limited release of results and reducing identifiers through key coding, have limited applicability in the clinic.44
In a clinical setting, there may be a tension between the goal of providing good health care to the patient and their family over a patient’s lifetime, and the need to protect individual privacy and confidentiality. A patient might become identifiable through whole-genome sequencing in which individuals and families with rare genomic disorders are cross-referenced between databases with different levels of access; genomic data generated from sequencing may be linked with nongenetic data allowing inferences to be made as to the data source and by directly linking genome sequences or variants to individual patient records for direct access.
Developing systems and processes that take into consideration individual confidentiality and minimize the risks of unanticipated data disclosure is important and there are a number of ways this can be achieved. These include restricting user access on grounds of necessity and proportionality (role-based access) or through data-access committees.44 However, the evolving role and responsibilities of bioinformaticians who process identifiable sequencing data need to be addressed, and questions remain about the scope and duty of health professionals to share relevant data with other at-risk family members. The extent and nature of data that can be accessed by and disclosed to different parties are subjects that are much debated, particularly in relation to variants that are unrelated to a patient’s clinical condition, phenotype, or known family history (often described as incidental findings).45,46
Concerns about the potentially harmful consequences of identification raise the issue of whether special protection should be placed on the storage of genomic data, especially whole-genome sequences,42 particularly as regulatory and professional responsibilities arise if sequence data is viewed as personal sensitive data under data-protection legislation. There are also strong arguments against this type of “genetic exceptionalism,”47,48 whereby clinical genomic data is given no additional special treatment above and beyond other forms of sensitive medical data created and held by health services.
The release of genome sequences to consumers who have purchased genome sequencing on a direct-to-consumer basis has generated debates about whether patients should be able to access to their own clinical data, including whole-genome data. There are also calls for the technological infrastructure to be put in place to enable patients to play a more active role in managing access to their own clinical data; indeed, participant-centric initiatives are becoming increasingly widespread in biomedical research.49 This was a recommendation of the US President’s Council of Advisors on Science and Technology in their report on health information technology50 and in the European Guidance on Data Protection.51 The use of these participant-centric initiatives in the research arena may be a precursor to wider clinical adoption.
The central role of informatics in clinical genome sequencing highlights the ambiguous regulatory position of algorithms within the current EU framework, since they are neither products nor medical devices and do not fall clearly under existing EU directives.52 Also unresolved is the extent to which increasing reliance on automation within the diagnostic process might influence professional liability for wrongful diagnosis and treatment, as well as missed diagnoses resulting in preventable conditions.
Conclusion
NGS technologies are already being used to aid the diagnosis of many inherited diseases, and the utility of whole-exome sequencing for clinical applications has been demonstrated.53,54,55,56 However, analysis and interpretation of genome data are complex processes and give rise to a number of issues and challenges that have to be overcome.57,58,59 Clinical implementation of NGS technologies will require standardization and integration of analysis pipelines and databases, and appropriate informatics support to facilitate medical decision making. These will require investment in information technology, informatics infrastructure, personnel, and training within health services, policy development regarding data sharing and privacy, and the establishment of a robust and centrally managed evidence base for clinical interpretation of genomic variants. Although there is no doubt that high-throughput genome sequencing technologies have the potential to benefit health, the development of informatics pipelines within an appropriate framework is essential for their responsible and effective translation into clinical practice.
Disclosure
The authors declare no conflict of interest.
References
Sboner A, Mu XJ, Greenbaum D, Auerbach RK, Gerstein MB . The real cost of sequencing: higher than you think! Genome Biol 2011;12:125.
Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ . Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet 2011;56:406–414.
Moorthie S, Mattocks C, Wright C . Review of massively parallel DNA sequencing technologies. The HUGO J 2011;5:1–12.
Flicek P, Birney E . Sense from sequence reads: methods for alignment and assembly. Nat Methods 2009;6(11 Suppl):S6–S12.
Kuhlenbäumer G, Hullmann J, Appenzeller S . Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat 2011;32:144–151.
Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods 2007;4:903–905.
Brockman W, Alvarez P, Young S, et al. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res 2008;18:763–770.
Li H, Ruan J, Durbin R . Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008;18:1851–1858.
The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–1073.
The International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 2010;467:52–58.
He D, Choi A, Pipatsrisawat K, Darwiche A, Eskin E . Optimal algorithms for haplotype assembly from whole-genome sequence data. Bioinformatics 2010;26:i183–i190.
Li Y, Willer C, Sanna S, Abecasis G . Genotype imputation. Annu Rev Genomics Hum Genet 2009;10:387–406.
Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 2008;40:722–729.
Stein L . Genome annotation: from sequence to biology. Nat Rev Genet 2001;2:493–503.
Lindblad-Toh K, Garber M, Zuk O, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011;478:476–482.
Myers RM, Stamatoyannopoulos J, Snyder M, et al. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011;9:e1001046.
The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007;447:799–816.
Harrow J, Denoeud F, Frankish A, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol 2006;7 Suppl 1:S4.1–S4.9.
Manolio TA, Brooks LD, Collins FS . A HapMap harvest of insights into the genetics of common disease. J Clin Invest 2008;118:1590–1605.
Magi A, Benelli M, Gozzini A, Girolami F, Torricelli F, Brandi M . Bioinformatics for next generation sequencing data. Genes 2010;1:294–307.
Pop M, Salzberg SL . Bioinformatics challenges of new sequencing technology. Trends Genet 2008;24:142–149.
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R . Dindel: accurate indel calls from short-read data. Genome Res 2011;21:961–973.
Ding L, Wendl MC, Koboldt DC, Mardis ER . Analysis of next-generation genomic data in cancer: accomplishments and challenges. Hum Mol Genet 2010;19(R2):R188–R196.
Flicek P, Amode MR, Barrell D, et al. Ensembl 2011. Nucleic Acids Res 2011;39(Database issue):D800–D806.
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006.
Schaaf CP, Wiszniewska J, Beaudet AL . Copy number and SNP arrays in clinical diagnostics. Annu Rev Genomics Hum Genet 2011;12:25–51.
Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–846.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009;84:524–533.
Cooper GM, Shendure J . Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 2011;12:628–640.
Kuntzer J, Eggle D, Klostermann S, Burtscher H . Human variation databases. Database 2010; doi:10.1093/database/baq015.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A . Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20: 110–121.
Huang N, Lee I, Marcotte EM, Hurles ME . Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 2010;6:e1001154.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Ng PC, Henikoff S . Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet 2006;7:61–80.
Vitkup D, Sander C, Church GM . The amino-acid mutational spectrum of human genetic disease. Genome Biol 2003;4:R72.
Eyre TA, Ducluzeau F, Sneddon TP, Povey S, Bruford EA, Lush MJ . The HUGO Gene Nomenclature Database, 2006 updates. Nucleic Acids Res 2006;34(Database issue):D319–D321.
The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet 2000;25:25–29.
Robinson PN, Mundlos S . The human phenotype ontology. Clin Genet 2010;77:525–534.
Kohonen-Corish MR, Al-Aama JY, Auerbach AD, et al.; Human Variome Project Meeting. How to catch all those mutations–the report of the third Human Variome Project Meeting, UNESCO Paris, May 2010. Hum Mutat 2010;31:1374–1381.
Sintchenko V, Coiera E . Developing decision support systems in clinical bioinformatics. Methods Mol Med 2008;141:331–351.
Kawamoto K, Lobach DF, Willard HF, Ginsburg GS . A national clinical decision support infrastructure to enable the widespread and consistent practice of genomic and personalized medicine. BMC Med Inform Decis Mak 2009;9:17.
McGuire AL, Fisher R, Cusenza P, et al. Confidentiality, privacy, and security of genetic and genomic test information in electronic health records: points to consider. Genet Med 2008;10:495–499.
Royal College of Physicians, Royal College of Pathologists and British Society for Human Genetics. Consent and confidentiality in clinical genetic practice: guidance on genetic testing and sharing genetic information. 2nd edn. Report of the Joint Committee on Medical Genetics. RCP, RCPath: London, 2011.
Lowrance WW, Collins FS . Ethics. Identifiability in genomic research. Science 2007;317:600–602.
Wolf SM, Lawrenz FP, Nelson CA, et al. Managing incidental findings in human subjects research: analysis and recommendations. J Law Med Ethics 2008;36:211, 219–48.
Wolf SM, Crock BN, Van Ness B, et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet Med 2012;14:361–384.
Evans JP, Burke W . Genetic exceptionalism. Too much of a good thing? Genet Med 2008;10:500–501.
Green MJ, Botkin JR . “Genetic exceptionalism” in medicine: clarifying the differences between genetic and nongenetic tests. Ann Intern Med 2003;138:571–575.
Kaye J, Curren L, Anderson N, et al. From patients to partners: participant-centric initiatives in biomedical research. Nat Rev Genet 2012;13:371–376.
President’s Council of Advisors on Science and Technology. Realising the Full Potential of Health Information Technology to Improve Healthcare for Americans: The Path Forward 2010.
Data Protection Working Group. Article 29, Working Document on the processing of personal data relating to health in electronic health records (HER).
European Commission. Medical devices: guidance document – Qualification and Classification of stand alone software.
Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 2010; 362:1181–1191.
Roach JC, Glusman G, Smit AF, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010;328:636–639.
Shearer AE, DeLuca AP, Hildebrand MS, et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci USA 2010;107:21104–21109.
Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med 2011; 3:87re3.
Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet 2010;375:1525–1535.
Guttmacher AE, McGuire AL, Ponder B, Stefánsson K . Personalized genomic information: preparing for the future of genetic medicine. Nat Rev Genet 2010;11:161–165.
Ormond KE, Wheeler MT, Hudgins L, et al. Challenges in the clinical application of whole-genome sequencing. Lancet 2010;375:1749–1751.
Acknowledgements
The content of this article forms part of a PHG Foundation Report on the implications of whole-genome sequencing for health (downloadable at www.phgfoundation.org). We acknowledge expert input from Andrew Devereau, Paul Flicek, and Timothy Hubbard and thank the project team at the PHG Foundation for their invaluable feedback on this work. The PHG Foundation is the working name of the Foundation for Genomics and Population Health, a charitable company registered in England and Wales (charity no. 1118664, company no. 5823194).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moorthie, S., Hall, A. & Wright, C. Informatics and clinical genome sequencing: opening the black box. Genet Med 15, 165–171 (2013). https://doi.org/10.1038/gim.2012.116
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.116
Keywords
This article is cited by
-
Development of the clinical next-generation sequencing industry in a shifting policy climate
Nature Biotechnology (2014)
