
Abstract
Purpose:
Copy number variants are an important source of human genome diversity. The widespread distribution of hemizygous copy number variants in the DNA of healthy humans suggests that haploinsufficiency is largely tolerated. However, little is known about the extent to which corresponding nullizygosity (two-copy deletion) is similarly tolerated.
Methods:
We analyzed a cohort of first cousin unions to enrich for shared parental hemizygous events and tested their Mendelian inheritance in offspring.
Results:
Analysis of autozygous DNA blocks (autozygome) in the offspring not only proved an efficient method of mapping “dispensable” DNA but also revealed potential selective bias against the occurrence of nullizygous changes. This bias was not restricted to genic copy number variants and was not accounted for by a high rate of miscarriages.
Conclusions:
The autozygome is an efficient way to map dispensable segments of DNA and may reveal selective bias against nullizygosity in healthy individuals.
Genet Med 2012:14(5):515–519
Similar content being viewed by others

Introduction
Recently, the long-held view of human genome structure was challenged by the exciting discovery of widespread variation in copy number that involves at least 5% of the human genome.1,2,3 Rapid technological development in array-based assays has helped speedup the discovery of more copy number variants (CNVs), with the emerging field of next-generation sequencing uncovering even more.4,5,6,7
However, this exponential increase of annotated CNVs has posed a challenge in defining what represents “benign” versus pathogenic structural variants. In particular, an increasingly relevant question is whether benign deletion CNVs in the hemizygous state remain so in the nullizygous state, especially when these CNVs are rare. Unfortunately, this question is difficult to address systematically because the relatively low frequency of many of these unique CNVs requires the screening of a prohibitive number of healthy individuals for detecting the CNV in question in the nullizygous state. The documentation of the nullizygous occurrence of CNVs in healthy individuals offers an opportunity to identify DNA segments that are “dispensable” and improve our understanding of the human genome.
The Saudi population is characterized by a high rate of consanguinity. With one-eighth of the genome of first cousins being shared on average (identical by descent), it is highly likely that some hemizygous CNVs will exist in these shared segments. Therefore, we have reasoned that an efficient alternative approach to the question raised above (i.e., what parts of DNA are dispensable) is to study the full set of autozygous intervals (autozygome) of the offspring of first cousin unions because their autozygomes are likely to be enriched for nullizygous CNVs and thus facilitating their rapid cataloging. In this study, not only were we able to demonstrate the effectiveness of this approach and the promise it holds when applied at a large scale, but we were also able to identify an apparent bias against nullizygosity for a subset of hemizygous CNVs, raising interesting possibilities about important biological roles played by these segments, most of which were non-genic in nature.
Materials and Methods
Human subjects
Subjects were recruited from multiplex Saudi families with parents who were first cousins, and written informed consent was obtained. For ethnically matched controls, we used healthy Saudi controls (n = 126) who were recruited for other projects in which they consented to having their DNA analyzed for other genetic studies ( Figure 1 ). This study was approved by the institutional review board of the King Faisal Specialist Hospital and Research Center.

Workflow diagram for the present study. CNV, copy number variant; IBD, identical by descent; ROH, runs of homozygosity.
Genotyping
Venous blood (5–10 ml) was collected in tubes containing EDTA from all subjects (families and controls). Following the manufacturer’s instructions, DNA extraction from EDTA tubes was performed using the Gentra DNA Extraction Kit (Qiagen, Germantown, MD). DNA samples were processed following the instructions provided by Affymetrix for their 6.0 platform (Affymetrix, Santa Clara, CA). In brief, genomic DNA was digested with StyI and then ligated to a common adaptor with T4 DNA ligase. After ligation, the template underwent PCR using Titanium Taq DNA polymerase (Clontech, Palo Alto, CA). Once the product had been purified, it was fragmented with DNAse I and end-labeled using terminal deoxynucleotidyl transferase, followed by a final step of target hybridization.
Bioinformatic analysis
CNV calling algorithm. We processed all .CEL files from Affymetrix 6.0 combined SNP and CNV microarrays using the Affymetrix Genotyping Console (GTC) 3.0 software for quality control, genotyping, copy number, and homozygosity analysis. Samples that did not yield quality control call rates of at least 94% were removed, as were those that had copy number median absolute pairwise difference (MAPD) ≥0.4. Shared parental hemizygous CNVs were defined as continuous stretches of at least five probes that had the same CNV call, were shared by both parents, and that existed on a haplotype background that was also shared by both parents in a given family. Region boundaries were defined from one probe upstream and one probe downstream of the probes with the common CNV calls.
Bias analysis
-
1
Assessment of transmission bias of shared parental hemizygous CNVs: According to the null hypothesis, shared parental hemizygous CNVs are expected to have a Mendelian distribution in the offspring compatible with 25:50:25 ratio for nullizygous:hemizygous:wild type. The extent and statistical significance of bias against nullizygosity was calculated using a χ2-test against an expected Mendelian proportion of number in the offspring.
-
2
In-depth characterization of shared parental hemizygous CNVs for which no corresponding nullizygous occurrence was observed in the offspring: These hemizygous CNVs were termed “biased”. Because the probability of observing nullizygosity for any shared parental hemizygous CNV among the offspring is 25%, it is possible that the biased segments represent only a statistical (insufficient number of events) rather than a biological phenomenon in the family study. Therefore, we set out to examine the allele frequency for these segments within the control cohort of 126 ethnically matched population samples. If a biased segment was identified in a nullizygous state in the control population, it was reclassified as “neutral”. Importantly, reclassifying a biased segment as neutral was based on >50% overlap with a nullizygous event observed in the healthy controls. Thus, we cannot exclude the possibility that some of those reclassified CNVs may in fact represent truly biased DNA segments. We then compared biased and neutral CNVs on the basis of length, conservation, and allele frequency ( Figure 3 ). For conservation analysis, 95 percentile of cross-species conservation rate (0–1) for each segment was collated from the phastCons 44-way alignment track from the UCSC Genome Browser database. Boxplots were generated in the R package to illustrate and compare all three parameters, and Idiographica 2.0 software (http://www.ncrna.org/idiographica) was used to show the distribution of the 140 events in the human genome.
Figure 3 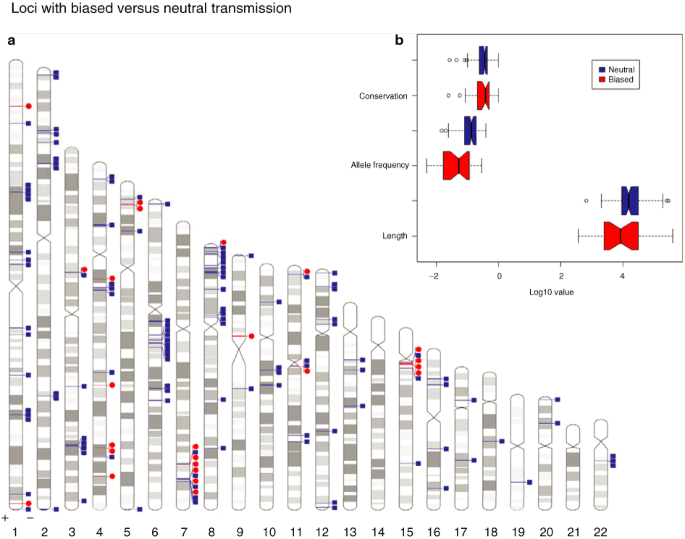
(a) Genome-wide distribution of copy number variants (CNVs) included in our analysis of 25 multiplex consanguineous families. “Biased” refers to shared parental hemizygous events that are never observed in nullizygous state in the children or the healthy population. “Neutral” refers to events that are observed at least once in a healthy individual. (b) Boxplot analysis comparing the two populations of CNVs (conservation, allele frequency, and length). Dashed lines and open circles denote standard deviation and outliers, respectively. Note that biased CNVs are rarer (P = 3.3 x 10−04) compared with neutral CNVs but were comparable in their length and degree of conservation.

Results
Autozygome is an efficient tool to map dispensable DNA
We enrolled 25 multiplex Saudi nuclear families (total 176 individuals) in which the parents were first cousins (average sibship size 5.0, range 2–14) and gave a written informed consent (see Supplementary Table S1 online). The entire sample set was run on Affymetrix 6.0 chip for genome-wide copy number analysis. We restricted our analysis to shared parental hemizygous CNVs and studied their Mendelian distribution in the offspring (the large number of children enhanced the power of this analysis). We were able to define a total of 176 shared parental events (average 7 per union, range 2–20), and we tested 853 transmission events. As expected by the nature of the shared parental hemizygous CNVs, their occurrence as nullizygous in the offspring was marked by a run of homozygosity resulting from the shared ancestral haplotype (identical by descent) that harbors the hemizygous CNV ( Figure 2 ).

A representative multiplex family with the inheritance pattern of two illustrative hemizygous copy number variants (CNVs; indicated by blue inverted triangle) that are shared by descent between the first cousin parents. Members of pedigree are color coded to match the output of the CNV calling algorithm shown below. Left: one CNV displaying Mendelian inheritance. Note that nullizygous children (cyan and purple) are displaying a run of homozygosity (thick color-matched horizontal bars) encompassing the CNV locus, indicating identical by descent (IBD). As expected, all other offspring (hemizygous and wild type) show no runs of homozygosity for that region. Right: another CNV exemplifying the bias against nullizygosity (all hemizygous except for orange, which is wild type).
Human DNA is apparently biased against nullizygosity
By comparing the observed distribution with the expected Mendelian 25:50:25 ratio (nullizygous:hemizygous:wild type), a highly significant bias against nullizygosity was identified (150:348:355, P < 5 × 10−28). This bias is unlikely to be the result of false calling of nullizygous CNVs in the offspring as hemizygous or wild type because only nullizygous CNVs would fall within runs of homozygosity. The presence of runs of homozygosity (as an indicator of identical by descent) was a filter that was used to streamline the detection of true nullizygous events because such events, by the design of this study, could not exist in a non–runs of homozygosity region. It is important to note here that although hemizygous CNVs can falsely appear as regions of homozygosity during data analysis, the extent of such homozygosity is limited by the boundaries of the CNV, a different pattern from the long runs of homozygosity that represents a true identical by descent segment surrounding the nullizygous CNV ( Figure 2 ). Indeed, polymerase chain reaction confirmation verified such regions to be truly nullizygous. Therefore, the observed bias against the occurrence of nullizygosity is unlikely to be explained solely on the basis of calling errors.
DNA segments with apparent bias against nullizygosity display less frequent variation in the general population
We then sought to further characterize the CNV events tested in the family study in a healthy Saudi cohort. First, we collapsed overlapping shared parental hemizygous CNVs between families to represent unique DNA segments (n = 140) and classified them as CNVs that are observed in nullizygous state (referred to as neutral, n = 79) and those that are not (biased, n = 61) in the family study. We then tested the frequency of these CNVs in a cohort of 126 Saudi controls run on the same Affymetrix 6.0 platform and found that 37 of the 61 biased CNVs are observed in the nullizygous state in the general population. Thus, the apparent bias in the family study for these DNA segments may have been a statistical rather than a biological phenomenon, i.e., nullizygosity could have been detected with a larger number of children. However, we cannot rule out a biological mechanism on the basis of linkage disequilibrium between these CNVs and recessive point mutations within the family in which the skewed Mendelian distribution was observed. Additive/combinatorial effect is another theoretical possibility (see below). The remaining 24 hemizygous CNVs were not observed in the healthy controls in the nullizygous state, and therefore, they retained the biased label (see Supplementary Table S2 online).
These events were then compared with the neutral CNVs for allele frequency, size, and degree of conservation. Interestingly, the only significant difference was in frequency where biased DNA segments showed much less frequent variation in the control population ( Figure 3 ). We caution that our reclassification of biased CNVs that are observed even once in the nullizygous state as neutral may have influenced this pattern. Importantly, 50% of these events did not contain any annotated gene, which raises the intriguing possibility of an important regulatory function of this non-genic DNA in human development.
No evidence of additive/combinatorial effect as the basis for the apparent bias against nullizygosity
Finally, we asked whether the bias could be explained on additive/combinatorial basis rather than recessive lethal basis. Under the additive/combinatorial model, the load of nullizygous events is what determines the degree of bias rather than the mere occurrence of a particular nullizygous event. To test for this, we reanalyzed the transmission patterns in the family study by limiting our analysis to shared parental hemizygous CNVs for which at least one corresponding nullizygous event was observed in the offspring. By doing that, we excluded CNVs that may have acted as recessive lethal, focusing our attention solely on the Mendelian transmission ratios for CNVs whose absence is clearly compatible with life. When we repeated the χ2-test using this subset of shared parental hemizygous CNVs, the result was complete loss of the bias. Therefore, the original bias likely originates from recessively acting rather than additive/combinatorial effects.
Discussion
The unique phenomenon of autozygosity lends itself in various ways to the improved annotation of the human genome.9 In this proof-of-concept study, we found that the autozygome can serve as an efficient tool for mapping dispensable regions of human DNA. “Dispensability” here refers to the compatibility with early human development. Indeed, nullizygosity for these segments may have adverse effects in the healthy offspring who carry these events but skipped detection because they are subtle, age dependent, or context dependent, i.e., may require interaction with other genetic factors in a complex fashion. The autozygome has also enabled us to identify some DNA segments that seem to be indispensable to normal human development despite being tolerated in a single-copy (hemizygous) state.
The assumption that inherited CNVs and those deposited in public databases as common structural variants are benign in nature has recently been brought into question.10 First, inherited CNVs can still be risk factors for disorders of complex genetics.11,12,13 Second, two recent reports describe clinical phenotypes associated with nullizygous loss of CNVs that are regarded as benign variants in the hemizygous state.14,15 Third, there is now a robust body of evidence in support of a model of enrichment rather than exclusive presence of some CNVs in patients with a variety of neurobehavioral phenotypes compared with controls. In other words, there is growing support of some CNVs in the general population being pathogenic but at reduced penetrance, perhaps even acting in a double-hit model.16,17,18 Therefore, the characterization of nullizygous deletions in a healthy cohort is of tremendous clinical utility because it defines DNA that is dispensable to normal human development.
This study showed that the normal human genome (including non-genic segments) can be biased against the occurrence of nullizygosity even when hemizygosity seems to be tolerated. One attractive explanation is that nullizygosity in some instances is not compatible with early embryogenesis because there was no history of recurrent miscarriages in the study families to denote incompatibility at a later stage. Although larger numbers of families would be needed, we note that this explanation is supported by the recent observation that a significant proportion of implantation failures may be caused by cytogenetic aberrations only discernible on comparative genomic hybridization.19
The exact mechanism through which nullizygosity for some part of the human DNA may adversely affect early human development and thus seem to be absent in healthy individuals is not clear, but several explanations, not mutually exclusive, may be proposed. The first obvious explanation is that these events may be acting simply as recessive lethal.8 However, our finding that some of these CNVs have a higher carrier frequency in the population than expected for lethal alleles suggests that this possibility may only apply in a subset of these CNVs (those with <1% population frequency), although heterozygote advantage or founder effect in the study population (as a result of tribal structure) cannot be excluded. Another possibility is that the bias is in fact against homozygosity for recessive lethal alleles that are in linkage disequilibrium with these CNVs rather than against the deletion itself. We propose that the two explanations are not mutually exclusive. One way to address the magnitude of the second possibility is exome sequencing to identify the presence of recessively acting lethal point mutations in linkage disequilibrium with the biased CNVs.
This study, to our knowledge, represents the first published attempt at cataloging dispensable DNA in humans. Because this is only a proof-of-concept study, we believe that a large-scale screening of healthy products of consanguineous unions will be an important follow-up study that can provide a much more comprehensive cataloging of dispensable DNA. Because the apparent bias observed against nullizygosity in some loci was not limited to genic CNVs, our data may provide yet another line of evidence supporting an important role for non-genic DNA in humans. It is worth noting here that the depletion of observed versus expected nullizygous events did not result in equal inflation of observed hemizygous and wild type bur rather a more significant inflation of the wild type. One possible explanation is that, these biased regions may have an important biological function that is dosage sensitive making wild type preferred over hemizygous occurrence. This observation will be the foundation for future and larger studies that systematically investigate this bias, particularly for non-genic DNA.
Disclosure
The authors declare no conflict of interest.
References
Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet 2004;36:949–951.
Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science 2004;305:525–528.
McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 2008;40:1166–1174.
Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 2009;41:1061–1067.
Mills RE, Walter K, Stewart C, et al.; 1000 Genomes Project. Mapping copy number variation by population-scale genome sequencing. Nature 2011;470:59–65.
Park H, Kim JI, Ju YS, et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 2010;42:400–405.
Sudmant PH, Kitzman JO, Antonacci F, et al.; 1000 Genomes Project. Diversity of human copy number variation and multicopy genes. Science 2010;330:641–646.
McConkey EH . Human Genetics: The Molecular Revolution: Jones & Bartlett Learning: Burlington, MA, 1993.
Alkuraya FS . Autozygome decoded. Genet Med 2010;12:765–771.
Buysse K, Delle Chiaie B, Van Coster R, et al. Challenges for CNV interpretation in clinical molecular karyotyping: lessons learned from a 1001 sample experience. Eur J Med Genet 2009;52:398–403.
Lee C, Scherer SW . The clinical context of copy number variation in the human genome. Expert Rev Mol Med 2010;12:e8.
Diskin SJ, Hou C, Glessner JT, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009;459:987–991.
Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008;82:477–488.
Curry CJ, Mao R, Aston E, et al. Homozygous deletions of a copy number change detected by array CGH: a new cause for mental retardation? Am J Med Genet A 2008;146A:1903–1910.
Knijnenburg J, Oberstein SA, Frei K, et al. A homozygous deletion of a normal variation locus in a patient with hearing loss from non-consanguineous parents. J Med Genet 2009;46:412–417.
Vorstman JA, van Daalen E, Jalali GR, et al. A double hit implicates DIAPH3 as an autism risk gene. Mol Psychiatry 2011;16:442–451.
Pinto D, Pagnamenta AT, Klei L, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010;466:368–372.
Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet 2010;6:e1000962.
Sher G, Keskintepe L, Keskintepe M, Maassarani G, Tortoriello D, Brody S . Genetic analysis of human embryos by metaphase comparative genomic hybridization (mCGH) improves efficiency of IVF by increasing embryo implantation rate and reducing multiple pregnancies and spontaneous miscarriages. Fertil Steril 2009;92:1886–1894.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(PDF 73 kb)
Supplementary Table S2
(PDF 92 kb)
Rights and permissions
About this article
Cite this article
Khalak, H., Wakil, S., Imtiaz, F. et al. Autozygome maps dispensable DNA and reveals potential selective bias against nullizygosity. Genet Med 14, 515–519 (2012). https://doi.org/10.1038/gim.2011.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2011.28
Keywords
This article is cited by
-
An exome-first approach to aid in the diagnosis of primary ciliary dyskinesia
Human Genetics (2020)
-
TLE6 mutation causes the earliest known human embryonic lethality
Genome Biology (2015)
-
Estimating the human mutation rate using autozygosity in a founder population
Nature Genetics (2012)
