
Abstract
Purpose: Congenital malformations are a major cause of morbidity and mortality in newborn infants, and genomic imbalances are a significant component of their etiology. The aim of this study was to evaluate the ability of prenatal multiplex ligation probe amplification screening to detect cryptic chromosomal imbalances in fetuses with ultrasound abnormalities of unknown etiology.
Methods: Multiplex ligation probe amplification was performed with three separate sets of probes: two for subtelomeric regions and one for mental retardation syndrome loci. Sixty-one fetuses with significant ultrasound anomalies and normal karyotype at a minimum of 400-band resolution were tested between January 2007 and January 2009.
Results: We identified four unbalanced rearrangements: one del 18pter/amp 5pter, one del 9pter, one 15q11q13 microdeletion, and one 22q11 microdeletion with atypical presentation. After genetic counseling, two of the pregnancies were terminated.
Conclusion: Multiplex ligation probe amplification analysis was able to identify clinically significant rearrangements in 6.5% of fetuses with prenatally identified sonographic abnormalities. This prospective study highlights that multiplex ligation probe amplification screening of fetuses with ultrasound abnormalities in the prenatal period is technically feasible and relevant for diagnosis and prognosis.
Similar content being viewed by others


Main
Congenital malformations are a major cause of morbidity and mortality in newborn infants and are diagnosed in about 3% of the population. Some congenital malformations are detected by ultrasound in the prenatal period. Standard karyotype obtained by chorionic villus sampling or amniocentesis reveals chromosomal aberrations in 18–35% of affected fetuses.1,2 To increase this detection rate, high-resolution methods based on fluorescence in situ hybridization (FISH), comparative genomic hybridization (metaphase spread or array-CGH) and quantitative multiplex polymerase chain reaction (PCR) techniques such as multiplex ligation probe amplification (MLPA) or quantitative multiplex PCR of short fragments have been applied but their use in prenatal diagnosis remains limited.
Telomere-specific FISH has already been shown to be a useful tool for detecting deletions and duplications in fetuses with major malformations.3,4 More recently, array-CGH analysis of fetal samples for the detection of small-copy-number DNA changes has been established as feasible.5–8 MLPA is a rapid, cost-effective method for screening for cryptic unbalanced subtelomeric rearrangements and interstitial imbalances located in regions associated with mental retardation (Mental Retardation Syndrome [MRS]-MLPA). Indeed, MLPA requires less than 2 days and MLPA results may be available in 3 days (in 4 days if a control FISH is required). To date, only one study has combined MRS-MLPA with subtelomeric MLPA, which improved the imbalance detection rate from 5.8% to 10.1% in children with mental retardation.9
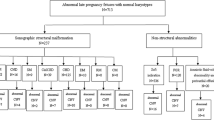
This study screened 61 fetuses with normal karyotype and ultrasound abnormalities suggestive of chromosomal aberration using subtelomeric MLPA and MRS-MLPA. To our knowledge, this is the first report of subtelomeric MLPA combined with MRS-MLPA used as a prenatal prospective screening method in the diagnostic evaluation of fetuses with ultrasound abnormalities.
MATERIALS AND METHODS
Patients
Sixty-one fetuses with one or more malformations suggestive of chromosomal abnormalities and normal karyotype were included in our prospective study during the period January 2007 to January 2009 (Table 1). For each fetus, a 400-band resolution karyotype with RHG and GTG banding was performed from amniotic fluid or chorionic villi. In cases of cardiac abnormalities, a 22q11.2 FISH was performed. When karyotype and FISH results were normal, we proposed to the patients to continue cytogenetic investigations by MLPA. All the 61 patients have accepted to participate at the study and an appropriate informed consent was obtained. The study was performed in full compliance with Declaration of Helsinki principles.
Multiplex ligation probe amplification
DNA was isolated from cultured chorionic villi or amniotic fluid cells using the NucleoSpin Blood kit (Macherey-Nagel) following the manufacturer's instructions. For chorionic villus samples, any maternal or anomalous material was removed by careful dissection under an inverted microscope to avoid maternal cell contamination. Three MLPA probe sets (MRC-Holland, Amsterdam, The Netherlands) were used. Two probe sets (P036 and P070) test all the subtelomeres except those of the acrocentric chromosome short arms (for these latter, probe recognition sequences are located on the q arm, in one of the first genes following the repeated sequences of the centromere). The simultaneous use of subtelomeric SALSA P036 and P070 probe sets improves the detection rate, as suggested by the manufacturer, and also helps to better interpret the results. In addition, the accuracy of subtelomeric MLPA was validated in a large patient cohort (n = 208) by Ahn et al.,10 who did not detect any false-negative and false-positive results. The third probe set (P245) tests a panel of 21 chromosomal regions involved in syndromes causing mental retardation. The regions covered by each kit are summarized in Table 1 and Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A104. All samples were tested with a combination of P036+P070 followed by P245 when negative.
One hundred twenty nanograms of DNA were used in the MLPA protocol according to the manufacturer's instructions. Reactions were performed on a TProfessional thermocycler (Biometra, Archamps, France). Three microliters of PCR products were analyzed by capillary electrophoresis on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Courtabeouf, France), and quantitative data were extracted using ABI Prism GeneScan Analysis and Genetyper software (Applied Biosystems). MLPA data analysis was performed using Microsoft Excel. One patient with an unbalanced translocation t(10;18)(qter;qter) and one patient with a 15q12 deletion were included as positive controls in each MLPA P036/P070 and P245 analysis, respectively, along with one negative control. Abnormal results were defined by a ratio above 1.3 for gain and below 0.7 for loss of genetic material.
Fluorescence in situ hybridization
Abnormal findings obtained by MLPA were validated by FISH on metaphase chromosomes according to standard procedures (Cytocell Technology or Vysis, Inc, Downers Grove, IL) using probes located in the suspected regions plus control probes. Peripheral blood lymphocytes from the parents were analyzed by FISH when a fetus tested was positive for rearrangement.
RESULTS
We detected cryptic imbalances in four of the 61 fetuses (6.5%) explored by MLPA. Two subtelomeric rearrangements and two pericentromeric microdeletions were identified. The clinical and cytogenetic data for these four cases are given below.
Patient A
The 24-year-old mother of this fetus had one healthy child and one miscarriage. Family history was nonsignificant. At 13 weeks of gestation, nuchal translucency was measured at 5.3 mm. Prenatal cytogenetic analysis of chorionic villi revealed a normal karyotype: 46,XX. An ultrasound performed at 18 weeks of gestation showed a suspected cardiac defect, and the patient underwent fetal echocardiography at 19 weeks of gestation, which revealed a complex cardiac defect with aortic valve agenesia and left ventricular dysfunction associated with poor prognosis. A 22q11.2 FISH was performed and was found to be normal. Further cytogenetic investigations were then conducted on the cryoconserved cells. Subtelomeric MLPA analysis revealed a loss of 18qter and a gain of 5pter (Fig. 1A), which were confirmed by FISH as an unbalanced translocation product (see Fig. A, Supplemental Digital Content 2, http://links.lww.com/GIM/A105). The minimal size of the 18qter deletion and of the 5pter amplification estimated by MLPA and FISH was 5.5 Mb and 367 Kb, respectively (CTDP1−, RP11-61A18−, RP11-504H5− for 18qter and PDCD6+ for 5pter). Karyotype and FISH analysis on metaphase spreads of the parents showed no aberrations. After genetic counseling, pregnancy was terminated at 20 weeks of gestation, in accordance with French legislation. Autopsy revealed severe maceration and partial liquefaction of the intra-abdominal organs, confirmed the complex heart defect, and revealed a single umbilical artery and bilateral pulmonary hypoplasia.

Gene dosage ratios for subtelomeric MLPA and MRS-MLPA. A, Dosage ratio of SALSA P036 (left) and P070 (right) probe sets for fetus A showing an amplification of the 5p telomere and a deletion of the 18q telomere. B, Dosage ratio of SALSA P036 (left) and P070 (right) probe sets for fetus B showing a deletion of the 9p telomere. C, Dosage ratio of SALSA P036 (left) and P070 (right) probe sets for fetus C showing a deletion of the 15q11.2 region. D, Dosage ratio of SALSA P245 probe sets for fetus D showing deletion of three probes targeting the 22q11.21 region.
Patient B
The 26-year-old mother of this fetus was pregnant with her second child. First-trimester ultrasound scan measured nuchal translucency thickness at 1.4 mm, and maternal serum screening gave a Down syndrome risk estimate of 1:840. Family history was nonsignificant. At 34 weeks of gestation, ultrasound examination revealed intrauterine growth retardation and hypospadias, prompting referral for amniocentesis. The karyotype, 46,XY, was normal, and FISH testing for 4p16.3 deletion (Wolf-Hirschhorn, OMIM194190) was negative. Subtelomeric MLPA analysis revealed a loss of 9pter (Fig. 1B). The deletion was confirmed by FISH (see Fig. B, Supplemental Digital Content 2, http://links.lww.com/GIM/A105). The minimal size estimated by MLPA and FISH was 810 Kb (DMRT1−, DOCK8−, RP11-143M15−). Karyotype and FISH analysis on metaphase spreads of the parents showed no aberrations. After counseling, and in accordance with French legislation, the parents decided to terminate the pregnancy at 38 weeks of gestation because of the high risk of mental retardation. The postmortem clinical assessment confirmed the hypospadias and showed a facial dysmorphism with upslanted palpebral fissures, microretrognatia, short neck, and widely spaced nipples.
Patient C
The patient was a 30-year-old gravida 2 para 1 with an unremarkable personal history. First-trimester ultrasound scan measured nuchal translucency thickness at 1.4 mm. Maternal serum screening gave a Down syndrome risk estimate of 1:1388. Ultrasound examination at 33 weeks of gestation revealed polyhydramnios associated with diminished fetal movements. Amniocentesis was performed for fetal karyotype and molecular investigations of myotonic dystrophy disease (MD, OMIM160900). The fetal karyotype (46,XX) was normal and genetic testing of the MD showed absence of increased number of the CTG repeat in the DMPK gene (n = 5–13). At 36 weeks of gestation, the subtelomeric MLPA detected a 15q11 microdeletion (Fig. 1C). The deletion was also confirmed by MRS-MLPA (data not shown). The minimal size estimated by the three sets of MLPA probes was 1.98 Mb (MKNR3- NDN-, SNRPN- UBE3 A-). The microdeletion was then validated by FISH (see Fig. C, Supplemental Digital Content 2, http://links.lww.com/GIM/A105). Karyotype and FISH analysis on metaphase spreads of the parents showed no aberrations. The parents decided to continue the pregnancy and delivery occurred at 39 weeks of gestation. The postnatal assessment showed hypotonia, poor sucking (requiring gavage feeding), small hands and feet with mild edema at the dorsum of feet, ogival palate, and facial dysmorphism typical of Prader-Willi syndrome (PWS, OMIM176270). Weight was 3080 g, length was 50 cm, and occipital frontal circumference was 36 cm.
Patient D
The patient was a 30-year-old primagravida. First-trimester nuchal translucency thickness was measured at 1.6 mm and maternal serum screening gave a low Down syndrome risk estimate of 1:2899. At 25 weeks of gestation, ultrasound examination revealed unilateral dysplasic multicystic kidney and bilateral club feet prompting referral for amniocentesis. Family history was nonsignificant, and parental renal ultrasounds were normal. The fetal karyotype, 46,XY, was normal. At 32 weeks of gestation, significant polyhydramnios was detected, and MLPA was performed on the cryoconserved cells. MRS-MLPA detected a 22q11.2 microdeletion (Fig. 1D) with a minimal size of 1.68 Mb (CLDN5-, GP1BB-, SNAP29-), which was confirmed by FISH (see Fig. D, Supplemental Digital Content 2, http://links.lww.com/GIM/A105). The parents refused to undergo their own karyotype testing and, after genetic counseling, decided to continue the pregnancy. The postnatal assessment at term confirmed the fetal abnormalities, and additional malformations were diagnosed. These included perimembranous interventricular communication, laryngeal stridor, and severe gastroesophageal reflux. Weight was 3540 g, length was 50.5 cm, and occipital frontal circumference was 36 cm.
DISCUSSION
The objective of this study was to evaluate the technical feasibility and utility of performing MLPA during the prenatal period. To our knowledge, this is the first study to assess MRS-MLPA combined with subtelomeric MLPA in the prenatal diagnostic screening of fetuses with abnormal ultrasound of unknown etiology.
We detected four submicroscopic rearrangements using three different MLPA probe sets in the 61 fetuses referred to our laboratory for abnormal ultrasound. These imbalances were confirmed by FISH on metaphase spreads.
Two subtelomeric rearrangements with clinical significance (del 18pter/amp 5pter and del 9pter) were detected (3.3%). In the postnatal period, subtelomeric rearrangements are detected in 3–7% of patients with congenital defects and/or mental retardation by FISH, MLPA, quantitative PCR and array-CGH.4,11–15 However, there are few reports concerning the detection of such imbalances in the prenatal period. Souter et al3 reported two prenatal cases of multiple fetal anomalies associated with subtle subtelomeric rearrangements detected by multi-subtelomere FISH. Gignac et al4 also screened 48 polymalformed fetuses with multi-subtelomere FISH and identified two clinically significant subtelomeric rearrangements (4%). More recently, Faas et al16 reported the detection of subtelomeric imbalance by FISH and MLPA in three fetuses presenting with ultrasound abnormalities. Their MLPA analysis was performed retrospectively in the postnatal period, and therefore had no influence on prenatal decision-making. The fetus B with the distal 9p deletion presented intrauterine growth retardation and hypospadias. The clinical phenotype of the 9p deletion syndrome is characterized by mental retardation, craniofacial dysmorphic features, notably trigonocephaly and a high frequency of genital and/or gonadal anomalies. Within this region the strongest candidates for the gonadal dysgenesis phenotype are the DMRT genes,17 which are included in the deletion found in fetus B.
We also detected two interstitial deletions (3.3%). Both subtelomeric MLPA and MRS-MLPA detected a 15q11q13 microdeletion associated with polyhydramnios and hypotonia. Recently, Bigi et al18 published the first report to describe a possible fetal phenotype in PWS, which could be recognizable after 30 weeks of gestation. They claimed that a particular position of hands and feet combined with diminished fetal movements and polyhydramnios is suggestive of PWS.18 However, fetus C did not present this kind of malposition of the extremities. Dudley and Muscatelli19 described a genotype-dependent variation in the obstetric characteristics of PWS, i.e., deletion or maternal uniparental disomy, making the fetal phenotype difficult to define. MRS-MLPA also detected a 22q11.2 microdeletion with atypical presentation. Renal dysplasia is frequently associated with 22q11.2 microdeletion, and there are multiple reports of polyhydramnios and club feet in this syndrome.20,21 However, to our knowledge, the prenatal association of unilateral dysplasic multicystic kidney, bilateral club feet, and polyhydramnios identified in fetus D has never been reported in 22q11.2 microdeletion syndrome. Taken together, these two cases suggest that MRS-MLPA may usefully detect microdeletion syndromes with atypical or incomplete prenatal presentation.
Array-CGH analysis has also been successfully applied for detection of chromosomal imbalances on fetal samples.5,7,8 However, arrays, which cover the whole genome at high resolution have identified a large number of copy number polymorphisms within the normal population.22 Thus, high-resolution array-CGH could detect copy number variations not directly associated with abnormal phenotype, and thus generate data of unknown significance.23 Hence, if array-CGH is to be used for prenatal diagnosis, the format of the array and the validation of results warrant careful consideration.6 Our results suggest that MLPA-based screening, particularly MRS-MLPA combined with subtelomeric MLPA, on fetuses with abnormal ultrasound would be an appropriate first-line routine diagnostic procedure when the karyotype is normal and echographic signs strongly suggest chromosomal abnormalities. Furthermore, MLPA is cheaper and less time-intensive than array-CGH, and it can analyze several patients in one run. In conclusion, we demonstrate the technical feasibility of MLPA on prenatal samples and describe for the first time the utility of subtelomeric MLPA and MRS-MLPA for prenatal diagnosis of cryptic chromosomal imbalances. Our results suggest that MLPA may be useful as a routine diagnostic procedure during the prenatal period. Cryptic rearrangements may be an under recognized cause of multiple fetal malformations but high-resolution genome-wide analyses are needed to evaluate their frequency. The prenatal applicability of this cost-effective technique gives a more precise prognosis and influences the perinatal management and care.
References
Eydoux P, Choiset A, Le Porrier N, et al. Chromosomal prenatal diagnosis: study of 936 cases of intrauterine abnormalities after ultrasound assessment. Prenat Diagn 1989; 9: 255–269.
Nicolaides KH, Snijders RJ, Gosden CM, Berry C, Campbell S . Ultrasonographically detectable markers of fetal chromosomal abnormalities. Lancet 1992; 340: 704–707.
Souter VL, Glass IA, Chapman DB, et al. Multiple fetal anomalies associated with subtle subtelomeric chromosomal rearrangements. Ultrasound Obstet Gynecol 2003; 21: 609–615.
Gignac J, Danis K, Tihy F, Lemyre E . Prenatal detection of subtelomeric rearrangements by multi-subtelomere FISH in a cohort of fetuses with major malformations. Am J Med Genet A 2006; 140: 2768–2775.
Le Caignec C, Boceno M, Saugier-Veber P, et al. Detection of genomic imbalances by array based comparative genomic hybridisation in fetuses with multiple malformations. J Med Genet 2005; 42: 121–128.
Rickman L, Fiegler H, Carter NP, Bobrow M . Prenatal diagnosis by array-CGH. Eur J Med Genet 2005; 48: 232–240.
Sahoo T, Cheung SW, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med 2006; 8: 719–727.
Vialard F, Molina Gomes D, Leroy B, et al. Array comparative genomic hybridization in prenatal diagnosis: another experience. Fetal Diagn Ther 2009; 25: 277–284.
Kirchhoff M, Bisgaard AM, Bryndorf T, Gerdes T . MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams-Beuren syndrome regions. Eur J Med Genet 2007; 50: 33–42.
Ahn JW, Mackie Ogilvie C, Welch A, et al. Detection of subtelomere imbalance using MLPA: validation, development of an analysis protocol, and application in a diagnostic centre. BMC Med Genet 2007; 8: 9.
De Vries BB, Winter R, Schinzel A, van Ravenswaaij-Arts C . Telomeres: a diagnosis at the end of the chromosomes. J Med Genet 2003; 40: 385–398.
Flint J, Knight S . The use of telomere probes to investigate submicroscopic rearrangements associated with mental retardation. Curr Opin Genet Dev 2003; 13: 310–316.
Rooms L, Reyniers E, Wuyts W, et al. Multiplex ligation-dependent probe amplification to detect subtelomeric rearrangements in routine diagnostics. Clin Genet 2006; 69: 58–64.
Shao L, Shaw CA, Lu XY, et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: a study of 5,380 cases. Am J Med Genet A 2008; 146A: 2242–2251.
Auber B, Bruemmer V, Zoll B, et al. Identification of subtelomeric genomic imbalances and breakpoint mapping with quantitative PCR in 296 individuals with congenital defects and/or mental retardation. Mol Cytogenet 2009; 2: 10.
Faas BH, Nillesen W, Vermeer S, et al. Detection of cryptic subtelomeric imbalances in fetuses with ultrasound abnormalities. Eur J Med Genet 2008; 51: 511–519.
Barbaro M, Balsamo A, Anderlid BM, et al. Characterization of deletions at 9p affecting the candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur J Hum Genet 2009; 17: 1439–1447.
Bigi N, Faure JM, Coubes C, et al. Prader-Willi syndrome: is there a recognizable fetal phenotype?. Prenat Diagn 2008; 28: 796–799.
Dudley O, Muscatelli F . Clinical evidence of intrauterine disturbance in Prader-Willi syndrome, a genetically imprinted neurodevelopmental disorder. Early Hum Dev 2007; 83: 471–478.
Kobrynski LJ, Sullivan KE . Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet 2007; 370: 1443–1452.
Devriendt K, Van Schoubroeck D, Eyskens B, et al. Polyhydramnios as a prenatal symptom of the digeorge/velo-cardio-facial syndrome. Prenat Diagn 1998; 18: 68–72.
Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature 2006; 444: 444–454.
Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Acknowledgements
We thank all the patients for their kind cooperation. We thank Dr Hélène Laurichesse, Prof. Denis Gallot, and Prof. Didier Lemery for providing the prenatal samples. We also thank Dr. Isabelle Cloix and Dr. Corinne Souquière for providing clinical data on the newborns, and Dr. Anne-Marie Beaufrère and Prof. Pierre Dechelotte for providing the postmortem clinical assessments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
Goumy, C., Gouas, L., Pebrel-Richard, C. et al. Prenatal detection of cryptic rearrangements by multiplex ligation probe amplification in fetuses with ultrasound abnormalities. Genet Med 12, 376–380 (2010). https://doi.org/10.1097/GIM.0b013e3181e074c6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181e074c6
Keywords
This article is cited by
-
First Successful Pregnancy After Pre-implantation Genetic Diagnosis by FISH for an Inversion Together with a Cryptic Translocation in India
Journal of Fetal Medicine (2016)
-
The Future of Prenatal Cytogenetics: From Copy Number Variations to Non-invasive Prenatal Testing
Current Genetic Medicine Reports (2013)
