
Abstract
Abstract: The skeletal dysplasias (osteochondrodysplasias) are a heterogeneous group of more than 350 disorders frequently associated with orthopedic complications and varying degrees of dwarfism or short stature. These disorders are diagnosed based on radiographic, clinical, and molecular criteria. The molecular mechanisms have been elucidated in many of these disorders providing for improved clinical diagnosis and reproductive choices for affected individuals and their families. An increasing variety of medical and surgical treatment options can be offered to affected individuals to try to improve their quality of life and lifespan.
Similar content being viewed by others


Main
Generalized disorders of cartilage and bone have been referred to as skeletal dysplasias, whereas those that affect an individual bone or group of bones have been referred to as dysostoses; however, these distinctions are blurring as their basic defects are elucidated. The skeletal dysplasias are associated with abnormalities in the patterning, development, maintenance, and size of the appendicular and axial skeleton and frequently result in disproportionate short stature. Until the early 1960s, most individuals with short stature were considered to have pituitary dwarfism, achondroplasia (short-limb dwarfism), or Morquio disease (short-trunked dwarfism). Presently, there are more than 350 well-characterized skeletal dysplasias that are classified primarily on the basis of clinical, radiographic, and molecular criteria.1 They result from mutations in various families of genes that encode extracellular matrix proteins, transcription factors, tumor suppressors, signal transducers (ligands, receptors, and channel proteins), enzymes, cellular transporters, chaperones, intracellular binding proteins, RNA processing molecules, cilia and cytoplasmic proteins, and a number of gene products of currently unknown function.
The skeletal dysplasias
The skeletal dysplasias are disorders associated with a generalized abnormality in the skeleton. Although each skeletal dysplasia is relatively rare, collectively the birth incidence of these disorders is almost 1/5000.2 These disorders range in severity from precocious arthropathy in relatively average stature individuals to severe dwarfism with perinatal mortality. These disorders can be associated with a variety of orthopedic, neurologic, auditory, visual, pulmonary, cardiac, renal, and psychological complications.
Embryology
The human skeleton (from the greek, skeletos, “dried up”) is a complex organ consisting of 206 bones (126 appendicular, 74 axial, and 6 ossicles). The musculoskeletal system also includes tendons, ligaments, and muscles, and, in addition to cartilage and bone, is involved in linear growth, mechanical support, movement, a blood cell and mineral reservoir, and protection of vital organs. These tissues and adipocytes all derive from mesenchymal precursor cells.
The patterning and architecture of the skeleton during fetal development determine the number, size, and the shape of the future skeletal elements.3 Uncondensed mesenchyme undergoes cellular condensations at sites of future bones and joints and this occurs by two mechanisms. In the process, mesenchymal cells differentiate into chondrocytes to form the cartilage anlagen and then, the center of the anlagen degrades, mineralizes, and is removed by osteoclast-like cells.4–8 This process spreads up and down the bones, allowing for vascular invasion and influx of osteoprogenitor cells. The periosteum in the midshaft region produces osteoblasts, which then synthesize the cortex.9 This is known as the primary ossification center. At the end of the cartilage anlagen, a similar process leading to the removal of cartilage, initiation of joint formation and a secondary ossification center forms, leaving a portion of cartilage model “trapped” between the expanding primary and secondary ossification center. This area is referred to as a cartilage growth plate or physis. There are three chondrocyte cell types in the growth plate: reserve/resting, proliferative, and hypertrophic. These growth plate chondrocytes undergo a tightly regulated program of proliferation, hypertrophy, degradation, and then replacement by bone (primary spongiosa) (Fig. 1, A).4 This is the major mechanism of skeletogenesis and is the mechanism by which bones increase in length and the articular surfaces increase in diameter. In contrast, the flat bones of the cranial vault and part of the clavicles and pubis formed by intramembranous ossification, where fibrous tissue, derived from mesenchymal cells, differentiates directly into osteoblasts which then directly lay down bone.10,11 These processes are under specific and direct genetic control.12 Chondrocytes produce a variety of proteins that compose the extracellular matrix. Some of the most prominent extracellular structural matrix proteins are the collagens, single molecules that associate into chains to form a triple helical structure. In the triple helix, every third amino acid is a glycine residue, and the general chain structure is denoted as Gly-X-Y, where X and Y are commonly proline and hydroxyproline. The helical structure undergoes numerous posttranslational modifications before its localization to the extracellular matrix where multiple triple helical chains become a fibril. The collagen helix can be composed of identical chains (homotrimeric), as in type II collagen, or can consist of different collagen chains (heterotrimeric), as seen in types I, IX, and XI collagen.13

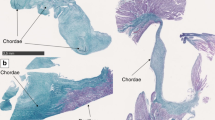
A, Normal growth plate morphology. Proliferating chondrocytes undergo hypertrophy, then apoptosis to become the primary spongiosum of bone. B, Abnormal growth plate in a case of metatropic dysplasia. Hypertrophic chondrocytes irregularly extend into the primary spongiosum, disturbing the normal architecture (arrow). C, Growth plate chondrocytes from a patient with diastrophic dysplasia demonstrating characteristic rings around the chondrocytes (arrow). P, proliferating chondrocyte; H, hypertrophic chondrocyte; B, bone; PS, primary spongiosum.
Collagens are widely distributed throughout the body and are expressed in a tissue specific manner. Collagens are further classified by the structures they form in the extracellular matrix. The most abundant collagens are the fibrillar types (I, II, III, V, and XI) and their extensive cross-linking provides mechanical strength that is necessary for high stress tissues such as cartilage, bone, and skin. Another group of collagens are the fibril associated collagens with interrupted triple helices and include collagen types IX, XII, XIV, and XVI. These collagens interact with fibrillar collagens and other extracellular molecules, including aggrecan, cartilage oligomeric matrix protein (COMP), decorin, fibulin, and numerous other sulfated proteoglycans.14 Collagen types VIII and X are nonfibrillar, short chain collagens; type X collagen is the most abundant extracellular matrix molecule expressed by hypertrophic chondrocytes during endochondral ossification.15 Mutations in genes that encode these collagens result in various skeletal dysplasias and highlight the importance of these molecules in skeletal development.
In the 1970s, there was increasing recognition of the genetic and clinical heterogeneity of these disorders and a new awareness of their complexity. There have been multiple attempts to classify these disorders, so that clinicians and scientists could effectively diagnose them and determine their pathogenicity (International Nomenclature of Constitutional Diseases of Bone, 1970, 1977, 1983, 1992, 2001, 2005, and 2009).1 The initial categories were purely descriptive and clinically based. With the recent explosion in determining the genetic basis of these diseases, the classification has evolved into a one that combines the older clinical and radiographic description with one that recognizes families of disorders that share a common molecular basis or pathway. The most recent updated classification can be found at www.isds.ch and listed in Table 1 are the skeletal dysplasias for which the underlying gene defect is known (this list is not inclusive of all genetic disorders with effects on the skeleton). The most widely used method for differentiating the skeletal disorders has been through the detection of skeletal radiographic abnormalities. Radiographic classifications are based on the different parts of the long bones that are abnormal (epiphyses, metaphyses, diaphyses) (Fig. 2). The epiphyseal, metaphyseal, and diaphyseal disorders can be further differentiated depending on whether the spine is involved (spondyloepiphyseal, spondylometaphyseal dysplasias [SMDs], or spondyloepimetaphyseal dysplasias [SEMDs]). The skeletal dysplasias can be also be differentiated into distinct disorders based on a variety of other clinical and radiographic findings.

Cartoon demonstrating the different portions of the appendicular skeleton that manifest radiographic abnormalities that aid in the clinical classification of the skeletal dysplasias.
Clinical evaluation and features in the chondrodysplasias
The skeletal dysplasias are generalized disorders of the skeleton, which usually result in disproportionate short stature. Most individuals with disproportionate short stature have skeletal dysplasias, and those with proportionate short stature have endocrine, nutritional, or other genetic or teratogenic disorders, although there are exceptions to this generalization. Some forms of osteogenesis imperfecta (OI) and hypophosphatasia can be associated with relatively normal body proportions.
A disproportionate body habitus may not be immediately apparent on physical examination. Therefore, anthropometric measurements such as upper/lower segment (U/L) ratio, sitting height, and arm span must be measured when evaluating a patient with short stature. Sitting height is an accurate measure of head and trunk length, but it requires special equipment for precise measurements. The lower segment is measured from the symphysis pubis to the floor at the inside of the heel. The upper segment is measured by subtracting the lower segment measurement from the total height. Upper and lower segment measurements can be obtained in a standing or supine position. These anthropometric ratios change with age as in a newborn the limbs are relatively short compared with the trunk. For example, a Caucasian newborn has an U/L ratio of about 1.7, a child between the ages of 8 and 10 years has a U/L segment ratio of ∼1.0, and an U/L segment ratio of 0.95 as an adult. Individuals presenting with disproportionate short stature will have altered ratios depending on whether they have short limbs, short trunk, or both. For example, an individual with short limbs and relatively normal trunk will have an increased U/L segment ratio and an individual with relatively normal limbs but short trunk will have a diminished U/L segment ratio. Another means of determining if there is disproportion is based on arm span measurements that are very close to total height in an average proportioned individual.
As in any disorder that has a genetic basis, it is critical to obtain an accurate family history, and this should include a history of other affected individuals and parental consanguinity. The skeletal dysplasias are genetically heterogenous and can be inherited as autosomal dominant, autosomal recessive, X-linked recessive, and X-linked dominant disorders, and rarer genetic mechanisms of disease including chromosomal deletions/duplications, germline mosaicism, and uniparental disomy have been seen.16–20 Furthermore, there is intrafamilial and interfamilial variability in many of these disorders. For many patients and families, accurate diagnosis and recurrence risk can have significant impact on their reproductive decisions. Another consideration for patients with short stature is that there is frequently nonassortative-mating, which can lead to compound phenotypes.21 For example, homozygous achondroplasia is lethal,22 and many newborns who inherit two different dominant mutations from parents with different disorders (compound heterozygotes) often are severely affected with combined features of both disorders. It is also important to obtain an accurate history as to the time of onset of short stature and whether it was present prenatally or not noticed until age 2 or 3 years. Of the 300+ skeletal dysplasias, ∼100 have onset in the prenatal period. For many of the other disorders, disproportionate short stature or orthopedic complications do not manifest until childhood.
If the limbs are involved, one should determine which segment is primarily involved: upper segment (rhizomelic: humerus and femur), middle segment (mesomelic: radius, ulna, tibia, and fibula), and distal segment (acromelic: hands and feet) because this can narrow the differential diagnosis. Numerous head and facial dysmorphisms are seen in the skeletal disorders. Affected individuals frequently have disproportionately large heads. Frontal bossing and flattened nasal bridge are characteristic of achondroplasia, the most common skeletal dysplasia.23 Cleft palate and micrognathia are commonly found in the types II and XI collagen abnormalities, and severe abnormally flattened midface with a turned up nose is frequently found in the chondrodysplasia punctata disorders, as well as others,24 whereas swollen ear pinnae in the neonatal period are seen in diastrophic dysplasia.25
Other organ systems, beyond the skeleton, can be involved, although not commonly. Congenital cardiac defects are seen in chondroectodermal dysplasia,26 the short-rib polydactyly disorders (complex outlet defects including isolated ventricular septal defects),27 and in Larsen syndrome.28 Gastrointestinal anomalies are rare among the skeletal disorders, but congenital megacolon can be seen in cartilage hair hypoplasia,29 malabsorption syndrome in Schwachmann-Diamond syndrome,30 and omphaloceles in otopalatodigital syndrome,31 and atelosteogenesis I/Boomerang dysplasia.32
Diagnosis and testing
After obtaining a thorough family history and physical examination, a full set of skeletal radiographs should be obtained including anteroposterior (AP), lateral and Towne views of the skull, AP and lateral views of the entire spine, and AP views of the pelvis and extremities, with separate AP views of the hands and feet. Occasionally, a lateral of knee can be helpful to diagnose the rare recessive form of megaepiphsyeal dysplasia (MED) associated with bilayered patella. Most of the important clues to diagnosis are in prepubertal skeletal radiographs. Once the epiphyses have fused to the metaphyses, determining the precise diagnosis can be exceedingly challenging. If an adult is evaluated, all attempts should be made to obtain any available childhood x-rays. There are many subtle clues in these skeletal radiographs that can lead to precise diagnosis. For example, punctuate calcifications in the areas of the epiphyses in the chondrodypsplasia punctata disorders, multiple ossification centers of the calcaneus seen in more than 20 disorders,33 and the type of hand shortening can aid in differentiating many disorders.
After obtaining radiographs, close attention should be paid to the specific parts of the skeleton (spine, limbs, pelvis, and skull) involved and to the location of the lesions (epiphyses, metaphyses, and spondylo-) (Figs. 3 and 4). As mentioned above, these radiographic abnormalities can change with age and if possible, radiographs spanning a time frame aids in diagnosis, since diagnosis sometimes cannot be made at an early age. It is critical to evaluate the cervical region (c-spine) in the skeletal dysplasias, especially in young children, because many are associated with distinct cervical vertebral abnormalities that need medical management (Table 2). Fractures can be seen in OI (all types), severe hypophosphatasia and the osteosclerotic (over-mineralizing) disorders including osteopetrosis (numerous forms). If a diagnosis cannot be reached after thorough evaluation, there are available resource such as the International Skeletal Dysplasia Registry (http://www.csmc/skeletaldysplasia) and the European Skeletal Dysplasia Network (http://www.esdn.org) that are available as consultants to provide diagnosis for these rare disorders.

Radiographs demonstrating abnormalities in the skeleton (pseudoachondroplasia). A, Lateral view of vertebral bodies showing rounded bodies with anterior beaking (arrow) and wide intervertebral disc spaces. B, A/P view of the knee showing irregular metaphyses (small arrow) and irregular, small for age epiphyses (large arrow). C, A/P view of the pelvis. Arrow points to small to almost absent, irregular epiphyses with mild metaphyseal abnormalities at the acetabulum surface and proximal femoral region.

Radiographs showing abnormalities (irregularities) in different portions of the skeleton that aid in the diagnosis. A, A/P view of the knee showing irregular metaphyses (arrow) in Metaphyseal Chondrodysplasia—Schmid type. B, A/P view of the pelvis with arrow pointing to very small distal epiphyses in Spondyloepiphyseal Dysplasia Congenita (SEDC). C, A/P view of the knee demonstrating very widened diaphysis in Diaphyseal Dysplasia–Camurati-Hunermann type. D, A/P view of the hand showing shortened metacarpals, phalanges with widened metaphysis (arrow) in acrodysostosis. E, A/P and lateral views of the spine illustrating irregular vertebral margins in Spondyloepiphyseal Dysplasia Tarda.
Morphologic studies of chondro-osseous tissue have revealed specific abnormalities in many of the skeletal dysplasias.34–37 In these disorders, histologic evaluation of chondro-osseous morphology can aid in making an accurate diagnosis, and absence of histopathologic alterations can rule out diagnoses. These studies need to be performed on cartilage growth plate, and while commonly performed on perinatal lethal skeletal disorders at time of autopsy, obtaining growth plate histology on individuals with nonlethal disorders can be done during elective surgeries. If affected individuals (children) are undergoing elective surgery, an iliac crest biopsy can be evaluated. Histomorphologic studies have also led to important insights into the pathogenesis of these disorders. On morphologic grounds, the chondrodysplasias can be broadly classified into those disorders (i) that have a qualitative abnormality in endochondral ossification (e.g., thanatophoric dysplasia—very short, irregular growth plate), (ii) have abnormalities in cellular morphology (e.g., atelosteogenesis—multinucleated giant cells), (iii) have abnormalities in matrix morphology (e.g., achondrogenesis IB—rings around the chondrocytes) (Fig. 1, C), and (iv) those in which the abnormality is primarily localized to the area of chondro-osseous transformation (e.g., metatropic dysplasia—cartilage trapped with the primary spongiosum) (Fig. 1, B), in contrast to those disorders with a relatively normal appearing growth plate (e.g., achondroplasia). Thus, histomorphologic studies can be valuable diagnostic aids and provide insights into the mechanisms producing disease.
There has been significant progress in the identification of the molecular defects in these disorders. Molecular diagnosis can be used to confirm a clinical and radiographic diagnosis, predict carrier status in those families at risk for a recessive disorder, and allow for prenatal diagnosis of at-risk fetuses. Because these are rare disorders, commercial testing is not always readily available; however, GeneTests (www.genetests.org), a publically funded website, provides current information on these diseases and those laboratories providing clinical genetic testing and those who are interested in material for ongoing research.
Management and treatment
The optimal management of this diverse set of disorders requires an understanding of their medical and psychosocial complications. This is often best accomplished by centers that have a multidisciplinary approach, which includes adult and pediatric physicians, as well as a group of orthopedists, rheumatologists, otolaryngologists, neurologists, neurosurgeons, ophthalmologists, and obstetrician-gynecologists who are experienced and committed to the care of these patients.
Many of the complications in these disorders result from skeletal abnormalities. Each disorder has its own characteristic set of complications. Shortening of the skull base in achondroplasia leads to a small foramen magnum, which can compress the spinal cord, especially in a flexed position and foramenotomy may be indicated. In those disorders associated with significant odontoid hypoplasia or cervical abnormalities such as Morquio disease, type II collagenopathies, SMD Kozlowski type, metatropic dysplasia, Larsen syndrome, and SEMD corner fracture type, flexion-extension films of the cervical spine should be monitored at regular intervals to assess for C1-C2 subluxation, especially in childhood. Moreover, recent evaluation of the cervical spine in many other skeletal disorders (Table 2) has increased the awareness of the unappreciated frequency of C1-C2 instability as well as other abnormalities (odontoid hypoplasia), and many experts now suggest cervical evaluation in all young children who are diagnosed with a skeletal dysplasia. If there is evidence for subluxation and/or cord compression, surgery for C1-C2 fixation is indicated. Genu varum (bow-legs) or genu valgum (knock-knees) are common in many skeletal disorders. This may cause knee or ankle pain and may require osteotomies. Children and adults with skeletal dysplasias should have regular eye and hearing examinations because certain disorders are associated with myopia, vitreal or retinal degeneration, glaucoma, and hearing loss. Speech, occupational, and physical therapy may also be indicated based the patient's needs.
Frequently, patients with these disorders have significant joint pain and/or joint limitation. In some disorders, such as the type II collagenopathies, pseudoachondroplasia, and MED, little cartilage remains at the knee or hips by adulthood that joint replacement is indicated for pain relief. Finally, weight control in adults with short stature is an ongoing issue and contributes to inactivity and adult-onset diseases such as diabetes, hypertension, and coronary disease.38,39
Chondrodysplasias
Achondroplasia
Achondroplasia is the most common of the nonlethal skeletal dysplasias (approximate frequency 1/20,000). The majority of these individuals has normal intelligence, a normal lifespan, and lead independent and productive lives. The mean final height in achondroplasia is 130 cm for men and 125 cm for women, and specific growth charts have been developed to document and track their linear growth, head circumference, and weight.40–42
In early infancy, there is the potential for serious compression of the cervicomedullary spinal cord secondary to a narrow foramen magnum, cervical canal, or both.43–48 Clinically, these infants can have central or obstructive sleep apnea, profound hypotonia, motor delay or excessive sweating, and can awake from sleep with emesis. Magnetic resonance imaging with flow studies in flexion47and extension are necessary to document the cerebrospinal fluid obstruction, which can require decompressive surgery.49 Other complications include nasal obstruction, thoracolumbar kyphosis, and occasionally hydrocephaly.49 From early childhood, individuals with achondroplasia can develop several orthopedic manifestations that include progressive bowing of the legs, lumbar lordosis, and hip flexion contractures. Recurrent ear infections can lead to chronic serous otitis media and deafness. Tympanic membrane tube placement is indicated in many of these patients. Craniofacial abnormalities can lead to dental malocclusion and tongue thrusting. As adults, the main potential medical complication is spinal canal narrowing, which can manifest by lower limb paresthesias, claudication, clonus, and bladder or bowel dysfunction. It is critical that these complaints are addressed promptly since without appropriate decompression surgery, paraplegia can result.49 Guidelines for the management of children with achondroplasia have been set forth by the American Academy of Pediatrics.50
Growth hormone has not been effective in increasing height in this disorder.51 Surgical limb lengthening has been used successfully to increase limb length by up to 12 inches,52 but patients need to recognize that this technique should be done during the teen years and is performed over a 2-year period. It is associated with numerous complications including stiff joints, increased fracture rate, and nerve damage.53 It is controversial as to whether young children should be subjected to this procedure. Many advocates of short stature individuals believe that the affected individual should have the maturity and independence to determine if they desire limb lengthening, and further this choice should not be parental in origin. Throughout their lives, individuals with achondroplasia and other skeletal dysplasias and their families experience various psychosocial challenges.54 This can be addressed by specialized medical and social support systems. Interactions with advocacy groups such as Little People of America (LPA: http://www.lpaonline.org) can provide both emotional support and medical information.
Osteogenesis imperfecta
OI is a heritable disorder of bone characterized by hypomineralization of the skeleton and was the first disorder hypothesized by McKusick to be due to a defect in collagen.55 OI is a common (approximate frequency 1/20,000) generalized disorder of connective tissue that predominantly affects bone.56
Four types of OI were delineated by Sillence57 in the late 1970s. However, a number of other types of OI have recently been recognized. They will be discussed below as mild, severe, and perinatal, which is somewhat arbitrary, because there is a broad spectrum of phenotypic variability.
Mild OI (known as OI type I)
Affected individuals with OI type I usually have a relatively mild clinical course, skeletal deformity, and radiologic appearance of the skeleton although there can be marked inter- and intrafamilial variability. They also account for the majority of individuals with OI. Individuals are usually short for their age relative to peers or to their unaffected family members but are not as severely affected as those individuals with other OI types. Many of these individuals experience numerous fractures, especially in childhood, and it is not uncommon for them to have as many as 20 fractures by the age of five. Their sclerae are blue that becomes gray to pale blue in adulthood. Arcus senilis, not related to lipid abnormalities, may occur in some patients. Other reported ocular defects include scleromalalcia, keratoconus, and retinal detachment.58 In some families, the teeth may manifest dentinogenesis imperfecta (DI) due to abnormal dentin. The deciduous and permanent teeth may have an opalescent and translucent appearance, which tends to darken with age.
During the second and third decades of life, a characteristic high-frequency sensorineural or mixed hearing loss can be detected.59,60 Many patients complain of easy bruising and this may result from the effects of the mutation on both skin and the vessels. Mildly affected patients may not have fractures at birth, although there is occasionally a fracture of a clavicle or extremity during delivery. Radiographically, affected newborns usually have wormian bones seen on lateral views of the skull, with significant osteopenia seen throughout the skeleton, especially the spine.61 Characteristically, the fracture rate falls dramatically after puberty, only to increase after menopause in females. Measurement of bone mineral density (amount of matter per square centimeter of bone) of the lumbar spine and hip, by dual-energy x-ray absorptometry at any age discloses a significant decrease in bone mass,59 although normative values are not available for young children. T scores (i.e., standard deviation from the young-adult mean BMD) are frequently in the range of −2.5 to −4.0 at the lumbar spine or proximal femur, consistent with the diagnosis of osteoporosis.
Severe deforming OI (known as OI types III and IV)
The deforming variety of OI encompasses a broad clinical spectrum. Similar to perinatal lethal OI, most cases are inherited as autosomal dominant (or de novo), although recurrent cases with autosomal recessive inheritance have been recently demonstrated. This form of OI is characterized by severe deformity of the limbs and marked kyphoscoliosis, thoracic deformity, and significant short stature. The extent of growth retardation is remarkable and in many adults height may not surpass 3 feet (90 to 100 cm). Abnormal cranial molding occurs in utero and during infancy, producing frontal bossing and a characteristic triangular shaped facies. Radiographically, wormian bones and delayed closure of the fontanelles may be observed well into the first decade of life.
Pulmonary function can be diminished because of distortion of the spine and thorax and can progress over time and lead to restrictive lung disease and sleep apnea. Unfortunately, because of diminished vital capacity, pulmonary insufficiency is a leading cause of death in subjects with severe OI. Many patients with scoliosis >60° develop respiratory compromise. Platybasia due to soft bones at the base of the skull may cause the external ear canals to slant upward as the base of the skull sinks on the cervical vertebrae. This may lead to communicating or obstructive hydrocephalus, cranial nerve palsies, and upper and lower motor neuron lesions. Headache, diplopia, nystagmus, cranial nerve neuralgia, decline in motor function, urinary dysfunction, and respiratory compromise are complications of basilar invagination.62,63 As opposed to blue sclera in milder OI, most severely affected OI patients have white sclera as adults. Approximately 25% of these patients have DI, necessitating constant dental care throughout and beyond childhood. Severe hearing impairment occurs in 10% of patients, although milder degrees of hearing loss are more common.
These patients have significant osteopenia, leading to multiple fractures in the upper and lower extremities and vertebral bodies, particularly before puberty. Unlike mild OI in which fractures tend to heal without deformity, fractures in severe OI frequently lead to skeletal deformity. Radiographs of the skeleton reveal marked osteopenia, thinning of cortical bone, narrowing of the diaphysis, and occasionally widening of the metaphysis, which merges into a dysplastic epiphyseal zone filled with whorls of partially calcified cartilage (i.e., popcorn deformity). Osteoporosis leads to collapse of vertebral end plates, contributing to worsening kyphoscoliosis. Pectus excavatum or pectus carinatum adds to thoracic deformity. In addition, lack of weight-bearing increases the severity of osteoporosis and increases the risk of fracture. Many individuals with the severe end of progressive deforming OI become wheel-chair bound at an early age or walk with mechanical assistance.
Perinatal lethal OI (known as OI type II)
Approximately 10% of OI patients have the severe neonatal form of the disease, lethal OI. Most cases result from sporadic (dominant) mutations64 in one of the type I procollagen genes; however, recently autosomal recessive forms of the disease have been documented.65–67 These infants present with severe bone fragility, multiple intrauterine fractures at various stages of healing, deformed extremities, and occasionally hydrops fetalis. Radiographic features include wormian bones, multiple fractures, crumbled bones, and characteristic beading of the ribs due to healing callus formation. It seems that one of the radiographic differences in the dominant versus recessive forms of perinatal lethal OI is the presences of broad, beaded ribs in the dominant form and thin, fractured ribs in the recessive forms.
Very rare forms of OI have been described. OI type V, reported by 2000,68 is distinguished by autosomal dominant inheritance, moderate fracture history, characteristic hyperplastic callus formation, limitation in forearm pronation, and supination due to intramembraneous bone formation at the joint, normal sclera, and no DI. Bone biopsy showed a mesh-like appearance of irregularly spaced lamellae, different from the woven bone seen in the severe forms of OI. The etiology of this rare form has not been established, but it does not result from mutations COL1A1(I) or COL1A2(I) which are the responsible disease genes for most dominant forms of OI. Another rare form of OI has been delineated, (OI type VI),69,70 inherited as an autosomal recessive disease with severe progressing OI, no DI and on bone biopsy, the lamellae have a fish-scale appearance. The underlying genetic defect is unknown.
Over the years, there have been multiple attempts to treat OI with a variety of vitamins, hormones, and drugs, none of which has been successful. During the past 10 years, bisphosphonates administered parenterally or orally to children and adults has demonstrated favorable results. Intravenously administered pamidronate has been shown to lead increased bone mass, decreased skeletal pain, and decreased fracture incidence in children with severe OI.71 Dosage regimens in different series for children and adults have varied from 1 to 3 mg/kg, administered intravenously at 2- to 4-month intervals, and lower-dosage regimens also have been reported.72 In general, reports indicate a significant increase in bone mass in children and a decrease in fracture rate. The effect is most marked in the spine, where vertebral remodeling may improve vertebral height. The currently recommended treatment regimen includes the use of a bisphosphonate, with adequate calcium and vitamin D supplementation to avoid hypercalciuria and to maintain serum vitamin D levels within normal limits. The use of surgery to correct deformities and to facilitate weight-bearing has been the subject of several reviews.72 In addition, every child with OI benefits from appropriate rehabilitative therapy.73,74
Molecular abnormalities and families of bone dysplasias
Based on similarities in clinical, radiographic, and histomorphology, the skeletal dysplasias have been classified into bone dysplasia families thought to share common pathophysiologic mechanisms,75,76 originally proposed by Spranger. Almost 20 years later, many of the predictions that were made based on plain film radiology were confirmed by molecular analysis. In recent years, there has been an explosion in our understanding of the basic biology of these disorders using a candidate gene approach, linkage analysis, positional cloning, homozygosity mapping, copy number variation, and human/mouse synteny for identification of the disease genes. These gene discoveries have demonstrated that identical phenotypes may result from mutations in different genes that act through a similar pathway (e.g., Noggin and GDF5 producing multiple synostoses syndrome) and that the same gene can cause different phenotypes (FGFR3 producing hypochondroplasia and thanatophoric dysplasia).77–80 The International Nomenclature group attempted to classify these disorders in a single classification based on clinical, radiologic, histopathologic, molecular, and pathway criteria. However, this has resulted in an imperfect system as some families are based on secure molecular information and others are created on purely similar clinical and radiographic features, because the molecular defects are unknown or different molecular pathways lead to similar phenotypes. Some of the more common occurring skeletal dysplasias will be discussed below, especially because they relate to families of disorders with similar pathogenetic mechanisms. Furthermore, there are still many skeletal dysplasias for which the disease genes are unknown.
Defects in extracellular structural proteins
Types I, II, IX, X, XI collagens, COMP, aggrecan, and perlecan
Since type II collagen was found primarily in cartilage, the nucleus pulposus, and the vitreous of the eye, it was hypothesized that skeletal disorders with significant epiphyseal, spine, and eye abnormalities would result from defects in type II collagen. Indeed, type II collagen defects have been identified in a spectrum of disorders ranging from perinatally lethal conditions to those with only mild arthropathy: achondrogenesis II, hypochondrogenesis, spondyloepiphyseal dysplasia congenital, spondlyoepimetaphyseal dysplasia—Strudwick type, Kniest dysplasia, Stickler syndrome, spondyloperipheral dysplasia, Czech dysplasia—metatarsal type, and “precocious” familial arthopathy. These disorders are referred to as type II collagenopathies and they all result from heterozygosity for mutations in COL2A1.81,82 Biochemical analysis of cartilage derived from these individuals shows electrophoretically abnormal type II collagen.
Mutations that result in a substitution for a triple-helical glycine residue seem to be the most common type of mutation.83–85 There is some correlation between the location of the mutation and the disease phenotype. In spondyloepiphyseal dysplasia, the glycine substitutions are scattered throughout the molecule; however, in Kniest dysplasia, the defects are in the more amino-terminal end of the molecule.86,87 Stickler syndrome (Table 1) is genetically heterogenous and results from mutations in COL2A1 and COL11A1, and nonocular forms from defects in COL11A2.88–90 In Stickler syndrome, the COL2A1 and COL11A1 mutations tend to be nonsense mutations resulting in premature translation stop codons. Patients with COL11A1 mutations tend to have a more severe eye phenotype and hearing loss than patients with COL2A1 mutations.
Individuals heterozygous for various COL11A2 mutations have a nonocular form of Stickler syndrome, with is consistent with the absent expression of COL11A2 in the vitreous humor.91 Oto-spondylo-megaepiphsyeal dysplasia is a rare autosomal recessive disorder caused by loss of function mutations in COL11A2.91 This disorder has radiographic similarities to Kniest dysplasia but is associated with profound sensorineural hearing loss and lack of ocular involvement.
Spondylometaphyeal dysplasia, Schmid type results from heterozygosity for mutations in COL10A1 (type X collagen).92 This skeletal dysplasia is associated with mild-to-moderate short stature, waddling gait, and leg pain during childhood. The radiographic abnormalities include metaphyseal irregularities, mild spinal changes,93 and enlarged capital femoral epiphysis in early childhood.
Aggrecan
Aggrecan, the most abundant proteoglycan in cartilage, has been recently shown to produce two disorders. Heterozygosity for a null mutation leads to spondyloepipyseal dysplasia-Kimberly type. This has been reported in a single family who demonstrated mild proportionate short stature, no dysmorphism, and early-onset arthropathy.94 Recently, individuals with extreme short stature and facial dysmorphisms including midface hypoplasia and relative prognathism were identified with an autosomal recessive form of an aggrecan disorder. Radiographic abnormalities in affected individuals included vertebral clefts, metaphyseal flaring, epiphyseal irregularities, brachydactyly, and accessory carpal bones. This novel skeletal disorder, SEMD Aggrecan type results from homozygosity for a missense mutation, asp2267-to-asn (D2267N) in the C-type lectin domain within the G3 domain of the molecule.95
Cartilage oligomeric matrix protein
Heterozygosity for mutations in COMP produces both pseudoachondroplasia and multiple epiphyseal dysplasia.96 COMP is a member of the thrombospondin family of proteins and consists of both epidermal growth factor and calcium binding, calmodulin domains.97 In both pseudoachondroplasia and multiple epiphyseal dysplasia (MED), disease producing mutations occur in the calmodulin-like domain, with a minority in the globular carboxyl-terminal domain. Almost all well-documented cases of pseudoachondroplasia have a COMP mutation, whereas MED can be due to mutations in more than 5 other genes as well (COL9A1, COL9A2, COL9A3, Matrillin 3, and DTDST). Almost 50% of MED patients have disease due to mutations in yet unidentified gene(s).98
Perlecan
Heparan sulfate proteoglycans are major components of basement membranes and the molecule may be involved in the stabilization of other molecules, as well as being involved in cell-to-cell adhesion. Perlecan is a ubiquitous heparan sulfate proteoglycan and has both angiogenic and growth-promoting attributes, primarily by acting as a coreceptor for fibroblast growth factor, FGF2. Homozygosity or compound heterozgyosity for mutations in the perlecan gene lead to Schwartz Jampel type I99 and dyssegmental dysplasia, Silverman-Handmaker type.100 Schwartz-Jampel type I is an autosomal recessive disorder characterized by short stature, myotonic myopathy, joint contractures, blepharophimosis, unusual pinnae, myopia, and pectus carnitum. Characteristic radiographic abnormalities include flat vertebrae, occasional coronal clefts, short tubular bones with metaphyseal flaring, fragmented capital femoral epiphyses, and large epiphyses of the distal femora and proximal tibiae.101 The dyssegmental dysplasias, Silverman-Handmaker, and Rolland Desbuqouis types are autosomal recessive forms of neonatal short-limbed dwarfism in which vertebral segmentation defects and short, thick, bowed long bones are the prominent radiographic features. Clinically, unusual facies, short neck, narrow thorax, cleft palate, encephalocele, and reduced joint mobility are commonly seen. The milder form of the dyssegmental dysplasias, Rolland-Desbuquois type, is frequently associated with survival beyond the newborn period.102 Because mutations in the gene that encodes perlecan produces dyssegmental dysplasia, Silverman-Handmaker type, the possibility exists that the two disorders are allelic.
Type I collagen
The mildest form of OI results of mutations affecting the COL1A1(I) and COL1A2(I) polypeptide chains of type I collagen resulting in quantitative deficiencies of type I collagen. Cultured fibroblasts from individuals with mild OI synthesize low amounts (approximately one half) of the expected amounts of type I collagen. Many of the reported mutations are nonsense and frameshift mutations and predicted to lead to premature termination codons, although there are some exceptions.64,119,120 OI can be inherited from a parent, occur de novo, as a new dominant mutation, and recurrence in sibling has been described due to germline mosaicism. The biochemical abnormality in the more severe forms of OI is due to the synthesis and secretion of abnormal type I collagen due to mutations in the genes encoding COL1A1 and COL1A2.121 As a result, the amount of normal type I collagen in bone is low, much of the secreted collagen is abnormally overmodified. Bone collagen fibers are thinner then normal, and at the intracellular level, type I collagen in retained within dilated endoplasmic reticulum.
Single glycine substitutions in the Gly-X-Y triplet of either COL1A1 or COL1A2 lead to severe forms of OI, as do some small deletions, all producing severe effects on the triple helix. The recessive forms of OI, that produce severe forms of OI, account for a small number of these cases and result from mutations in the genes encoding either CRTAP (cartilage associated protein) or P3H1 (prolyl-3-hydroxylase 1) or cyclophilin B.106,122,123These molecules form a complex that hydroxylates the third position residue at proline 986 (Pro986) and mutations in this complex lead to underhydroxylation of that residue.
Defects in TRPV4 (calcium channel)
TRPV4 is a calcium-permeable ion channel of the vanilloid subfamily of TRP channels. The TRP superfamily consists of 28 cation channels that are key to ion homeostasis and are cellular sensors required for taste, vision, and detection of temperature and mechanical forces.103 Recently, heterozygosity for mutations in TRPV4 gene was shown to produce a broad spectrum of clinical disorders from autosomal dominant brachyolmia to SMD Kozlowski type to metatropic dysplasia.104 These mutations span the molecule with no clear genotype/phenotype correlations, but all the published mutations lead to increased intracellular calcium levels with in vitro stimulation,104,105 suggesting that these are activating mutations. However, it has also been shown that heterozygosity for mutations in TRPV4 produce congenital distal spinal muscle atrophy, scapuloperonal spinal muscle atrophy, and Charcot-Marie-Tooth disease type 2C106–108 demonstrating that mutations in TRPV4 can lead to nonskeletal dysplasia phenotypes.
Defects in intracellular structural proteins
Intracellular structural proteins are ubiquitously expressed, thus the finding that mutations in the genes encoding Filmain A, Filamin B, and the tubulin-specific chaperone E produced primarily skeletal and connective tissue disorders was surprising.109–113 The filamins are cytoskeleton proteins involved in multicellular processes including providing structure to the cell, facilitating signal transduction and transport of small solutes, allowing communication between the intracellular and extracellular environment, and participating in cell division and motility. Defects in these genes have a profound affect on the skeleton that includes the absence of bone formation to significant joint dislocations. The mechanism by which these mutations produce disease is still not clear. Absence of filamin B due to either compound heterozygosity or homozygosity for nonsense mutations produces spondylocarpotarsal syndrome.110 This disorder is characterized by progressive spinal fusions and carpal/tarsal fusions as well as joint laxity with other rare malformations.114 Heterozgyosity for filamin B defects, either missense or deletion mutations, results in a spectrum of disorders from the most radiographically severe, Boomerang dysplasia to Atelosteogenesis I/Atelosteogenesis III to Larsen syndrome.109,115 The severe end of the spectrum is frequently lethal, while most individuals with Larsen syndrome survive beyond infancy. Affected individuals can have extensive large joint dislocations leading to long-term joint complications, kyphoscliosis, cleft palate, and tracheal malformations.
Heterozygosity for Filamin A mutations, an X-liked gene, can produce a spectrum of disorders: frontometaphsyeal dysplasia, Melnick-Needles syndrome, otopalatodigital syndromes types I and II (OPDI and II), and periventriular nodular heterotopia.116,117 Some of these disorders such as Melnick-Needles syndrome and periventriular nodular heterotopia are inherited as X-linked dominant disorders and are frequently associated with early lethality in males. In contrast, the X-linked recessive disorders OPDI and II are seen predominantly in males. Frontometaphyseal dysplasia is inherited in an X-linked dominant disorder, where females are affected although less severely than their affected male offspring. Mutations in the Filamin A cluster in different regions of the molecule, the second calponin homology domain and among the repeat domains that surround the first hinge region.118
Summary
Although the osteochondrodysplasias are individually relatively rare disorders, affected individuals have significant complications throughout their lives, due to patterning defects, effects on linear growth, and finally, loss of normal structural cartilage as a cushion later in life. The 350 genetic disorders considered to be “osteochondrodysplasias” share phenotypic findings and molecular pathways, and an appreciation of their similarities and differences, underlying genetic etiology, and distinct complications is critical for appropriate medical care of these individuals. The explosion in delineating the molecular defects has shown the complexity of cartilage and bone as tissues and the large number of cellular processes necessary for a normal skeleton. We hope that the recent advances in the understanding of these disorders will lead to effective therapeutics to improve the quality of life for affected individuals.
References
Superti-Furga A, Unger S . Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A 2007; 143: 1–18.
Orioli IM, Castilla EE, Barbosa-Neto JG . The birth prevalence rates for the skeletal dysplasias. J Med Genet 1986; 23: 328–332.
Kornak U, Mundlos S . Genetic disorders of the skeleton: a developmental approach. Am J Hum Genet 2003; 73: 447–474.
Ballock RT, O'Keefe RJ . Physiology and pathophysiology of the growth plate. Birth Defects Res C Embryo Today 2003; 69: 123–143.
Ballock RT, O'Keefe RJ . The biology of the growth plate. J Bone Joint Surg Am 2003; 85: 715–726.
Ahmed YA, Tatarczuch L, Pagel CN, Davies HM, Mirams M, Mackie EJ . Physiological death of hypertrophic chondrocytes. Osteoarthritis Cartilage 2007; 15: 575–586.
Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M . Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 2008; 40: 46–62.
Yang Y . Skeletal morphogenesis during embryonic development. Crit Rev Eukaryot Gene Expr 2009; 19: 197–218.
Stains JP, Civitelli R . Cell-to-cell interactions in bone. Biochem Biophys Res Commun 2005; 328: 721–727.
Eames BF, Helms JA . Conserved molecular program regulating cranial and appendicular skeletogenesis. Dev Dyn 2004; 231: 4–13.
Eames BF, de la Fuente L, Helms JA . Molecular ontogeny of the skeleton. Birth Defects Res C Embryo Today 2003; 69: 93–101.
Colnot C . Cellular and molecular interactions regulating skeletogenesis. J Cell Biochem 2005; 95: 688–697.
Kuivaniemi H, Tromp G, Prockop DJ . Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat 1997; 9: 300–315.
Eyre DR . Collagens and cartilage matrix homeostasis. Clin Orthop Relat Res 2004;( 427 suppl): S118–S122.
Chan D, Ho MS, Cheah KS . Aberrant signal peptide cleavage of collagen X in Schmid metaphyseal chondrodysplasia. Implications for the molecular basis of the disease. J Biol Chem 2001; 276: 7992–7997.
Rimoin DL . Molecular defects in the chondrodysplasias. Am J Med Genet 1996; 63: 106–110.
Superti-Furga A, Bonafe L, Rimoin DL . Molecular-pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet 2001; 106: 282–293.
Edwards MJ, Wenstrup RJ, Byers PH, Cohn DH . Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a mutation in the COL1A2 gene of type I collagen. The mosaic parent exhibits phenotypic features of a mild form of the disease. Hum Mutat 1992; 1: 47–54.
Stevenson DA, Brothman AR, Chen Z, Bayrak-Toydemir P, Longo N . Paternal uniparental disomy of chromosome 14: confirmation of a clinically-recognizable phenotype. Am J Med Genet A 2004; 130: 88–91.
Stevenson DA, Bleyl SB, Maxwell T, Brothman AR, South ST . Mandibulofacial dysostosis in a patient with a de novo 2;17 translocation that disrupts the HOXD gene cluster. Am J Med Genet A 2007; 143A: 1053–1059.
Unger S, Korkko J, Krakow D, Lachman RS, Rimoin DL, Cohn DH . Double heterozygosity for pseudoachondroplasia and spondyloepiphyseal dysplasia congenita. Am J Med Genet 2001; 104: 140–146.
Pauli RM, Conroy MM, Langer LO Jr, et al. Homozygous achondroplasia with survival beyond infancy. Am J Med Genet 1983; 16: 459–473.
Hunter AG, Bankier A, Rogers JG, Sillence D, Scott CI Jr, Medical complications of achondroplasia: a multicentre patient review. J Med Genet 1998; 35: 705–712.
Sheffield LJ, Danks DM, Mayne V, Hutchinson AL . Chondrodysplasia punctata-23 cases of a mild and relatively common variety. J Pediatr 1976; 89: 916–923.
Lamy M, Maroteaux P . [Diastrophic nanism]. Presse Med 1960; 68: 1977–1980.
Piacentini G, Digilio MC, Sarkozy A, Placidi S, Dallapiccola B, Marino B . Genetics of congenital heart diseases in syndromic and non-syndromic patients: new advances and clinical implications. J Cardiovasc Med (Hagerstown) 2007; 8: 7–11.
Chen CP, Chang TY, Tzen CY, Lin CJ, Wang W . Sonographic detection of situs inversus, ventricular septal defect, and short-rib polydactyly syndrome type III (Verma-Naumoff) in a second-trimester fetus not known to be at risk. Ultrasound Obstet Gynecol 2002; 19: 629–631.
Kiel EA, Frias JL, Victorica BE . Cardiovascular manifestations in the Larsen syndrome. Pediatrics 1983; 71: 942–946.
Bacchetta J, Ranchin B, Brunet AS, et al. Autoimmune hypoparathyroidism in a 12-year-old girl with McKusick cartilage hair hypoplasia. Pediatr Nephrol 2009; 24: 2449–2453.
Khan S, Hinks J, Shorto J, Schwarz MJ, Sewell WA . Some cases of common variable immunodeficiency may be due to a mutation in the SBDS gene of Shwachman-Diamond syndrome. Clin Exp Immunol 2008; 151: 448–454.
Young K, Barth CK, Moore C, Weaver DD . Otopalatodigital syndrome type II associated with omphalocele: report of three cases. Am J Med Genet 1993; 45: 481–487.
Canki-Klain N, Stanescu V, Stanescu R, Sinkovec J, Debevec M, Maroteaux P . Lethal short limb dwarfism with dysmorphic face, omphalocele and severe ossification defect: Piepkorn syndrome or severe “boomerang dysplasia”?. Ann Genet 1992; 35: 129–133.
Cormier-Daire V, Savarirayan R, Unger S, Rimoin DL, Lachman RS . “Duplicate calcaneus”: a rare developmental defect observed in several skeletal dysplasias. Pediatr Radiol 2001; 31: 38–42.
Rimoin DL . The chondrodystrophies. Adv Hum Genet 1975; 5: 1–118.
Sillence DO, Horton WA, Rimoin DL . Morphologic studies in the skeletal dysplasias. Am J Pathol 1979; 96: 813–870.
Rimoin DL, Sillence DO . Chondro-osseous morphology and biochemistry in the skeletal dysplasias. Birth Defects Orig Artic Ser 1981; 17: 249–265.
Horton WA, Rimoin DL . Histochemical characterization of the endochondral growth plate: a new approach to the study of the chondrodystrophies. Birth Defects Orig Artic Ser 1978; 14: 81–93.
Hecht JT, Hood OJ, Schwartz RJ, Hennessey JC, Bernhardt BA, Horton WA . Obesity in achondroplasia. Am J Med Genet 1988; 31: 597–602.
Hoover-Fong JE, Schulze KJ, McGready J, Barnes H, Scott CI . Age-appropriate body mass index in children with achondroplasia: interpretation in relation to indexes of height. Am J Clin Nutr 2008; 88: 364–371.
Hoover-Fong JE, McGready J, Schulze KJ, Barnes H, Scott CI . Weight for age charts for children with achondroplasia. Am J Med Genet A 2007; 143A: 2227–2235.
Horton WA, Rotter JI, Kaitila I, et al. Growth curves in achondroplasia. Birth Defects Orig Artic Ser 1977; 13: 101–107.
Hunter AG, Hecht JT, Scott CI Jr, Standard weight for height curves in achondroplasia. Am J Med Genet 1996; 62: 255–261.
Nelson FW, Hecht JT, Horton WA, Butler IJ, Goldie WD, Miner M . Neurological basis of respiratory complications in achondroplasia. Ann Neurol 1988; 24: 89–93.
Hecht JT, Francomano CA, Horton WA, Annegers JF . Mortality in achondroplasia. Am J Hum Genet 1987; 41: 454–464.
Danielpour M, Wilcox WR, Alanay Y, Pressman BD, Rimoin DL . Dynamic cervicomedullary cord compression and alterations in cerebrospinal fluid dynamics in children with achondroplasia. Report of four cases. J Neurosurg 2007; 107( 6 suppl): 504–507.
Pauli RM, Scott CI, Wassman ER Jr, et al. Apnea and sudden unexpected death in infants with achondroplasia. J Pediatr 1984; 104: 342–348.
Pauli RM . Surgical intervention in achondroplasia. Am J Hum Genet 1995; 56: 1501–1502.
Pauli RM, Horton VK, Glinski LP, Reiser CA . Prospective assessment of risks for cervicomedullary-junction compression in infants with achondroplasia. Am J Hum Genet 1995; 56: 732–744.
Gordon N . The neurological complications of achondroplasia. Brain Dev 2000; 22: 3–7.
Trotter TL, Hall JG . Health supervision for children with achondroplasia. Pediatrics 2005; 116: 771–783.
Kanaka-Gantenbein C . Present status of the use of growth hormone in short children with bone diseases (diseases of the skeleton). J Pediatr Endocrinol Metab 2001; 14: 17–26.
Yasui N, Kawabata H, Kojimoto H, et al. Lengthening of the lower limbs in patients with achondroplasia and hypochondroplasia. Clin Orthop Relat Res 1997; 344: 298–306.
Venkatesh KP, Modi HN, Devmurari K, Yoon JY, Anupama BR, Song HR . Femoral lengthening in achondroplasia: magnitude of lengthening in relation to patterns of callus, stiffness of adjacent joints and fracture. J Bone Joint Surg Br 2009; 91: 1612–1617.
Hill V, Sahhar M, Aitken M, Savarirayan R, Metcalfe S . Experiences at the time of diagnosis of parents who have a child with a bone dysplasia resulting in short stature. Am J Med Genet A 2003; 122A: 100–107.
McKusick VA . Heritable disorders of connective tissue. V. Osteogenesis imperfecta. J Chronic Dis 1956; 3: 180–202.
Martin E, Shapiro JR . Osteogenesis imperfecta: epidemiology and pathophysiology. Curr Osteoporos Rep 2007; 5: 91–97.
Sillence DO . Osteogenesis imperfecta nosology and genetics. Ann N Y Acad Sci 1988; 543: 1–15.
Madigan WP, Wertz D, Cockerham GC, Thach AB . Retinal detachment in osteogenesis imperfecta. J Pediatr Ophthalmol Strabismus 1994; 31: 268–269.
Pedersen U . Osteogenesis imperfecta clinical features, hearing loss and stapedectomy. Biochemical, osteodensitometric, corneometric and histological aspects in comparison with otosclerosis. Acta Otolaryngol Suppl 1985; 415: 1–36.
Pedersen U, Melsen F, Elbrond O, Charles P . Histopathology of the stapes in osteogenesis imperfecta. J Laryngol Otol 1985; 99: 451–458.
Davie MW, Haddaway MJ . Bone mineral content and density in healthy subjects and in osteogenesis imperfecta. Arch Dis Child 1994; 70: 331–334.
Charnas LR, Marini JC . Communicating hydrocephalus, basilar invagination, and other neurologic features in osteogenesis imperfecta. Neurology 1993; 43: 2603–2608.
Charnas LR, Marini JC . Neurologic profile in osteogenesis imperfecta. Connect Tissue Res 1995; 31: S23–S26.
Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007; 28: 209–221.
Baldridge D, Schwarze U, Morello R, et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 2008; 29: 1435–1442.
Cabral WA, Chang W, Barnes AM, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007; 39: 359–365.
Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 2006; 355: 2757–2764.
Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 2000; 15: 1650–1658.
Glorieux FH, Ward LM, Rauch F, Lalic L, Roughley PJ, Travers R . Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res 2002; 17: 30–38.
Land C, Rauch F, Travers R, Glorieux FH . Osteogenesis imperfecta type VI in childhood and adolescence: effects of cyclical intravenous pamidronate treatment. Bone 2007; 40: 638–644.
Glorieux FH . Experience with bisphosphonates in osteogenesis imperfecta. Pediatrics 2007; 119( suppl 2): S163–S165.
Gonzalez E, Pavia C, Ros J, Villaronga M, Valls C, Escola J . Efficacy of low dose schedule pamidronate infusion in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab 2001; 14: 529–533.
Binder H, Conway A, Gerber LH . Rehabilitation approaches to children with osteogenesis imperfecta: a ten-year experience. Arch Phys Med Rehabil 1993; 74: 386–390.
Binder H, Conway A, Hason S, et al. Comprehensive rehabilitation of the child with osteogenesis imperfecta. Am J Med Genet 1993; 45: 265–269.
Spranger J . Pattern recognition in bone dysplasias. Prog Clin Biol Res 1985; 200: 315–342.
Spranger J . Bone dysplasia ‘families’. Pathol Immunopathol Res 1988; 7: 76–80.
Gong Y, Krakow D, Marcelino J, et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 1999; 21: 302–304.
Dawson K, Seeman P, Sebald E, et al. GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet 2006; 78: 708–712.
Wilcox WR, Tavormina PL, Krakow D, et al. Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. Am J Med Genet 1998; 78: 274–281.
Bellus GA, McIntosh I, Smith EA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 1995; 10: 357–359.
Horton WA . Molecular genetic basis of the human chondrodysplasias. Endocrinol Metab Clin North Am 1996; 25: 683–697.
Francomano CA, McIntosh I, Wilkin DJ . Bone dysplasias in man: molecular insights. Curr Opin Genet Dev 1996; 6: 301–308.
Korkko J, Cohn DH, Ala-Kokko L, Krakow D, Prockop DJ . Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis. Am J Med Genet 2000; 92: 95–100.
Lee B, Vissing H, Ramirez F, Rogers D, Rimoin D . Identification of the molecular defect in a family with spondyloepiphyseal dysplasia. Science 1989; 244: 978–980.
Nishimura G, Haga N, Kitoh H, et al. The phenotypic spectrum of COL2A1 mutations. Hum Mutat 2005; 26: 36–43.
Wilkin DJ, Artz AS, South S, et al. Small deletions in the type II collagen triple helix produce kniest dysplasia. Am J Med Genet 1999; 85: 105–112.
Winterpacht A, Hilbert M, Schwarze U, Mundlos S, Spranger J, Zabel BU . Kniest and Stickler dysplasia phenotypes caused by collagen type II gene (COL2A1) defect. Nat Genet 1993; 3: 323–326.
Williams CJ, Ganguly A, Considine E, et al. A-2→G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred. Am J Med Genet 1996; 63: 461–467.
Annunen S, Korkko J, Czarny M, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet 1999; 65: 974–983.
Sirko-Osadsa DA, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH . Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J Pediatr 1998; 132: 368–371.
Vikkula M, Mariman EC, Lui VC, et al. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995; 80: 431–437.
Warman ML, Abbott M, Apte SS, et al. A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat Genet 1993; 5: 79–82.
Savarirayan R, Cormier-Daire V, Lachman RS, Rimoin DL . Schmid type metaphyseal chondrodysplasia: a spondylometaphyseal dysplasia identical to the “Japanese” type. Pediatr Radiol 2000; 30: 460–463.
Gleghorn L, Ramesar R, Beighton P, Wallis G . A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet 2005; 77: 484–490.
Tompson SW, Merriman B, Funari VA, et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet 2009; 84: 72–79.
Briggs MD, Mortier GR, Cole WG, et al. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet 1998; 62: 311–319.
Adams JC . Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol 2001; 17: 25–51.
Chapman KL, Briggs MD, Mortier GR . Review: clinical variability and genetic heterogeneity in multiple epiphyseal dysplasia. Pediatr Pathol Mol Med 2003; 22: 53–75.
Arikawa-Hirasawa E, Le AH, Nishino I, et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am J Hum Genet 2002; 70: 1368–1375.
Arikawa-Hirasawa E, Wilcox WR, Le AH, et al. Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat Genet 2001; 27: 431–434.
Spranger J, Hall BD, Hane B, Srivastava A, Stevenson RE . Spectrum of Schwartz-Jampel syndrome includes micromelic chondrodysplasia, kyphomelic dysplasia, and Burton disease. Am J Med Genet 2000; 94: 287–295.
Aleck KA, Grix A, Clericuzio C, et al. Dyssegmental dysplasias: clinical, radiographic, and morphologic evidence of heterogeneity. Am J Med Genet 1987; 27: 295–312.
Clark K, Middelbeek J, van Leeuwen FN . Interplay between TRP channels and the cytoskeleton in health and disease. Eur J Cell Biol 2008; 87: 631–640.
Krakow D, Vriens J, Camacho N, et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet 2009; 84: 307–315.
Rock MJ, Prenen J, Funari VA, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet 2008; 40: 999–1003.
Auer-Grumbach M, Olschewski A, Papic L, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet 2010; 42: 160–164.
Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet 2010; 42: 165–169.
Landoure G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet 2010; 42: 170–174.
Farrington-Rock C, Firestein MH, Bicknell LS, et al. Mutations in two regions of FLNB result in atelosteogenesis I and III. Hum Mutat 2006; 27: 705–710.
Krakow D, Robertson SP, King LM, et al. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet 2004; 36: 405–410.
Robertson SP, Twigg SR, Sutherland-Smith AJ, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet 2003; 33: 487–491.
Robertson SP . Molecular pathology of filamin A: diverse phenotypes, many functions. Clin Dysmorphol 2004; 13: 123–131.
Parvari R, Hershkovitz E, Grossman N, et al. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet 2002; 32: 448–452.
Bicknell LS, Farrington-Rock C, Shafeghati Y, et al. A molecular and clinical study of Larsen syndrome caused by mutations in FLNB. J Med Genet 2007; 44: 89–98.
Bicknell LS, Morgan T, Bonafe L, et al. Mutations in FLNB cause boomerang dysplasia. J Med Genet 2005; 42: e43.
Robertson SP . Filamin A: phenotypic diversity. Curr Opin Genet Dev 2005; 15: 301–307.
Robertson SP . Filamin a, periventricular nodular heterotopia, and West syndrome. Epilepsia 2006; 47: 1082.
Robertson SP, Jenkins ZA, Morgan T, et al. Frontometaphyseal dysplasia: mutations in FLNA and phenotypic diversity. Am J Med Genet A 2006; 140: 1726–1736.
Willing MC, Cohn DH, Byers PH . Frameshift mutation near the 3′ end of the COL1A1 gene of type I collagen predicts an elongated Pro alpha 1(I) chain and results in osteogenesis imperfecta type I. J Clin Invest 1990; 85: 282–290.
Willing MC, Cohn DH, Starman B, Holbrook KA, Greenberg CR, Byers PH . Heterozygosity for a large deletion in the alpha 2(I) collagen gene has a dramatic effect on type I collagen secretion and produces perinatal lethal osteogenesis imperfecta. J Biol Chem 1988; 263: 8398–8404.
Byers PH . Brittle bones—fragile molecules: disorders of collagen gene structure and expression. Trends Genet 1990; 6: 293–300.
van Dijk FS, Nesbitt IM, Zwikstra EH, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009; 85: 521–527.
Van Dijk FS, Nesbitt IM, Nikkels PG, et al. CRTAP mutations in lethal and severe osteogenesis imperfecta: the importance of combining biochemical and molecular genetic analysis. Eur J Hum Genet 2009; 17: 1560–1569.
Acknowledgements
DK and DLR are supported by NIH HD22657 and DK is supported by NIH 1R01DE019567.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Krakow, D., Rimoin, D. The skeletal dysplasias. Genet Med 12, 327–341 (2010). https://doi.org/10.1097/GIM.0b013e3181daae9b
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181daae9b
This article is cited by
-
Circadian rhythm modulates endochondral bone formation via MTR1/AMPKβ1/BMAL1 signaling axis
Cell Death & Differentiation (2022)
-
De novo serine synthesis regulates chondrocyte proliferation during bone development and repair
Bone Research (2022)
-
Development and validation of an expanded targeted sequencing panel for non-invasive prenatal diagnosis of sporadic skeletal dysplasia
BMC Medical Genomics (2021)
-
Estimation of the carrier frequencies and proportions of potential patients by detecting causative gene variants associated with autosomal recessive bone dysplasia using a whole-genome reference panel of Japanese individuals
Human Genome Variation (2021)
-
Skeletal dysplasia-like syndromes in wild giraffe
BMC Research Notes (2020)
