
Abstract
Purpose: Hereditary hemochromatosis has not been fully evaluated in the non-Ashkenazi population and is considered to be relatively rare. After ascertaining three unrelated hereditary hemochromatosis families of North African Jewish origin with the HFE C282Y/C282Y genotype, we evaluated the C282Y and H63D allele frequencies among the different Jewish ethnic groups in Israel, in particular North African Jews.
Methods: Data were collected from three Israeli Medical Centers. North African, Oriental, Yemenite, and Sephardic Jewish healthy individuals were assessed for the C282Y and H63D alleles.
Results: The C282Y allele frequency was 1.02% (6/586 chromosomes), and the H63D allele frequency was 13.82% (81/586 chromosomes) in the North African Jewish group. The C282Y allele was not detected in the other non-Ashkenazi groups. The H63D allele frequency was 12.5% (38/304 chromosomes) in Oriental Jews, 14.9% (14/94 chromosomes) in Yemenite Jews, and 9.3% (11/118 chromosomes) in Sephardic Jews.
Discussion: Hereditary hemochromatosis is underrecognized among North African Jews, who have carrier frequencies of 1/58 and 1/4 for C282Y and H63D, respectively. HFE-hereditary hemochromatosis is not rare among this population as currently thought and merits increased awareness to prevent endpoint disease. The frequent occurrence of β-thalassemia trait and HFE-H63D in non-Ashkenazi Jews raises the possibility of genetic interactions contributing to iron overload when coinherited and requires further evaluation.
Similar content being viewed by others
Main
Hereditary hemochromatosis (HH; OMIM 235200), caused by a mutation in the HFE gene,1 is an autosomal recessive disorder characterized by inappropriately high absorption of iron by the gastrointestinal mucosa, resulting in excessive storage of iron, particularly in the liver, skin, pancreas, heart, joints, and testes. Two major HFE variants account for most HH cases: the major allele C282Y and the minor allele H63D,1,2 which present with variable frequencies in different populations.
Throughout history, Jewish ethnic groups were often isolated from the surrounding non-Jewish populations as a result of religious restrictions and cultural differences and from each other by geographic boundaries. Thus, their respective genetic loads differ because of new mutations that originated in the specific communities, including those acquired by admixture with the people they lived among. More ancient founder mutations that were inherited before the communities dispersed are common to all Jews.3 The major Jewish ethnic groups are the Ashkenazi, North African, Sephardic, Oriental, and Yemenite populations.4,5
Studies of the Jewish Ashkenazi population6 showed allele frequencies of 1.3 and 9.7% for the C282Y and H63D mutations, respectively, among 381 healthy controls. Only one study assessed these mutations in a non-Ashkenazi Jewish group consisting of 147 Sephardic subjects, but the North African and Oriental cohorts were too small—55 and 53 individuals, respectively—to draw conclusions.7 No ethnic group-specific stratification was performed in a study from Israel.9 This lack of data in addition to the absence of reports on endpoint disease in this population have resulted in an unclear view of HH among health care professionals and classification of the disease as relatively rare by the Official Genetic Database web site (Golden Helix—http://www.goldenhelix.org/israeli/).
We ascertained three HFE-HH unrelated Moroccan Jewish families with the C282Y/C282Y genotype. This led to a multicenter evaluation of C282Y and H63D allele frequencies among the Jewish ethnic groups in Israel, focusing on North African Jewry, which accounts for 14% of the total Israeli Jewish population (CBS, Statistical Abstract of Israel 2008, Central Bureau of Statistics).
MATERIALS AND METHODS
Participating centers
Data were collected from the three medical centers that performed the population study: Assaf Harofeh (Zerifin), Tel Aviv-Sourasky (Tel Aviv), and Shaare Zedek (Jerusalem).
Subjects
Data related to C282Y and H63D mutations were collected from healthy subjects from the following Jewish ethnic groups: Ashkenazi, North African (including Morocco, Libya, Algiers, and Egypt), Sephardic (including Italy, Greece, Turkey, and Bulgaria), Oriental (including Iraq, Iran, Egypt, Syria, Lebanon, Buckchara, and Kurdistan), and Yemenite. All participants were from homogeneous ethnic groups. Definition of origin was based on patient history. Informed consent was obtained from all participants, and the study was approved by the National Review Board.
Genetic analysis
Total genomic DNA was isolated from peripheral blood leukocytes or by buccal smear, as previously described,10 and was anonymously analyzed. The C282Y and H63D mutations were assessed using restriction fragment length polymorphism according to Feder et al.1 following the method of Lynas.11 The number of tested individuals and carriers from each origin was recorded for each mutation.
RESULTS
Ascertained families
Family 1
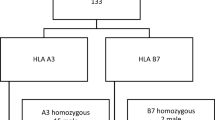
The index case was detected at the age of 57 years and was of Moroccan Jewish origin (Fig. 1, I-4). He presented with hypertension, type II diabetes mellitus, chronic renal failure, normocytic normochromic anemia, congestive heart failure, hepatomegaly, diffuse dark pigmentation of the skin, and pitting leg edema. Ferritin serum levels were >1000 μg/L, and transferrin saturation was 50%. Liver biopsy showed excessive iron overload with diffuse fibrosis, and genetic analysis showed homozygosity for the C282Y mutation. As noted in the pedigree, cascade genetic screening in this family disclosed additional family members: a homozygote for C282Y (Fig. 1, I-3), a compound heterozygote for C282Y/H63D (Fig. 1, II-2), and a nonrelated member of Libyan origin who was heterozygote for C282Y (Fig. 1, II-1). These family members were either not affected clinically or biochemically or not assessed. The brother of the index case (I-2) died at the age of 50 years because of cirrhosis without any previously defined risk factor, suggesting endpoint HFE-HH disease. A partial report of this family has been submitted to the European Journal of Internal Medicine as a case report.

Pedigrees of three families with genotypes.
Family 2
The index case was of Jewish Moroccan ancestry, referred at the age of 37 years (Fig. 1 II-8). He had arthritis of unknown origin, and biochemical studies showed ferritin level of 1145 μg/L, iron saturation of 70%, and elevated liver function tests (aspartate aminotransferase-49, alanine aminotransferase-85). HFE mutation analysis revealed a C282Y/C282Y genotype. Other homozygotes (Fig. 1, II-3, II-6, and II-10) or compound heterozygous individuals related to the family (Fig. 1, I-2, I-5, I-7, and I-9) were either not affected (clinically or biochemically) or not assessed.
Family 3
The index case was of Jewish Moroccan ancestry and was referred from the hematology clinic at the age of 15 years with increased ferritin level of 591 μg/L (Fig. 1, II-1). The parents refused genetic evaluation of their child. Paternal and maternal biochemical investigation revealed iron saturation of 61 and 28%, and genetic assessment demonstrated C282Y/C282Y and C282Y/WT genotype, respectively (Fig. 1, I-1 and I-2). Other family members were not assessed.
Population study
A total of 860 healthy individuals were evaluated in this study: 309 Ashkenazi Jews and 551 non-Ashkenazi Jews (Table 1). C282Y allele frequency among Ashkenazi Jews was 1.62% (10/618 chromosomes) and among non-Ashkenazi Jews was 0.54% (6/1102 chromosomes). When the latter group was further stratified, no C282Y was detected in the Oriental, Yemenite, and Sephardic groups, corresponding to 304, 94, and 118 evaluated chromosomes, respectively. In contrast, allele frequency of C282Y among North African Jews was 1.02% (6/586 chromosomes). The largest subgroup in the North African group was the Moroccan Jews (82%); of 478 screened chromosomes, 5 carried the C282Y genotype, yielding an allele frequency of 1.05% (95%CI, 0.15–1.95). The additional C282Y carrier, related by marriage to family 1 (II-1), was of Libyan Jewish origin and was not included in the total allele frequency calculation.
H63D allele frequency in Ashkenazi Jews was 14.08% (87/618 chromosomes), and in the non-Ashkenazi group was 13.1% (144/1102 chromosomes). Stratification according to ethnic groups demonstrated allele frequencies in the Oriental, Yemenite, and Sephardic groups of 12.5, 14.9, and 9.3% (38/304, 14/94, and 11/118 chromosomes), respectively. Allele frequency of H63D among the North African Jews was 13.82% (81/586 chromosomes). According to Pearson's χ2 test, the study groups were found to be in Hardy-Weinberg equilibrium.
DISCUSSION
We have presented here three unrelated families with HFE-HH, with endpoint disease in multiple individuals, all of Moroccan Jewish ancestry. When combined with the carrier frequency of ∼1/58 for the C282Y and ∼1/4 for H63D alleles detected in our cohort of 293 North African Jews (Table 1), our data suggest that HH may be underrecognized in this ethnic group. C282Y allele frequency in our Ashkenazi cohort of 309 controls is in accord with previous studies,6 reaching an allele frequency of 1.62% (Table 1).
Unlike the North African and the Ashkenazi groups, the C282Y allele was absent from the Oriental and Sephardic groups. Even when combined with data reported by Matas et al.,7 the allele frequency of 0.24% in both groups is in the rare range (Table 1). The Yemenite group investigated here for the first time exhibited an allele frequency of 0%.
In contrast to the rarity of the C282Y allele, the H63D allele was more frequent in all the ethnic groups, ranging from 9.3% in the Sephardic group to 14.08% in the Ashkenazi group, in accordance with previous studies, whereas the Yemenite group had an allele frequency of 14.89% (Table 1).
The rarity of the C282Y allele in the Oriental, Yemenite, and Sephardic Jewish groups (0–0.24%, Table 1), in contrast to the North African and Ashkenazi Jews where C282Y allele frequencies reach 1.15–1.38%, is suggestive of a gene flow resulting from a host influence. The C282Y allele frequency is null to low in Asia and Mediterranean basin,12–15 and in line with the Oriental, Yemenite, and Sephardic groups. In Spain, where Jews resided before their expulsion to North Africa, allele frequency ranges from 1.7–4.4%.14,16 Allele frequency of C282Y in Northern to Eastern Europe is relatively high, ranging from 6.3–14.7%,13,14 corresponding to that of the Ashkenazi Jews residing in that region. With regard to the H63D allele, which is frequent in all Jewish ethnic groups in the range of 11.5–14.89% (Table 1), this is compatible with the worldwide spread of this mutation: 30% in Spain and 8–9% in Saudi Arabia and North Africa.14 It may have resulted from a single event that occurred before the Jewish communities dispersed or from gene flow from independent events that took place in their respective host populations.17,18 These assumptions require further haplotype assessments.
Although penetrance for the characteristic clinical endpoints (diabetes mellitus, cirrhosis, and liver carcinoma) is estimated at 1% of C282Y homozygotes,19,20 a correlation with iron-overload-related disease has been documented in 28.4 and 1.2% of homozygous men and women, respectively.15 This underscores the need for increased awareness of this disorder among North African Jews, similar to that for Ashkenazi Jews, because phlebotomy or iron chelating can spare affected individuals from endpoint disease.21 This increased awareness is especially required in light of the repeated HH's late endpoint disease presentations as was presumed to occur in Family 1 (Fig. 1, I-2 and I-4).
Penetrance for biochemically documented iron overload is even higher, estimated at 40–70%.12,22 Although C282Y homozygosity confers the highest risk for iron overload, H63D mutation (in homozygosity or compound heterozygosity) was also associated with increased risk.7,23–27 Furthermore, aggravation of iron overload may occur when additional nongenetic or genetic modifier such as β-thalassemia trait is coinherited.27–32
The frequency of β-thalassemia trait among Oriental Jews originating from Kurdistan is high, with carrier frequency estimated at 20%. The disease is also relatively frequent among Jews from Middle Eastern countries and North Africa; the heterozygote frequency in these populations is ∼2–4% (Official Genetic Database web site).33,34 Given that the frequencies of the alleles in the Jewish population are in Hardy-Weinberg equilibrium, double heterozygosity for H63D and β-thalassemia is expected in more than 1/200 individuals from these ethnic groups, and homozygosity for the first, with heterozygosity for the second, is expected in more than 1/3200 individuals from these groups. In view of side effects associated with iron overload, such as hepatocellular carcinoma,35 other cancers,36 and immunodeficiency,37 iron body stores should be assessed and genetic evaluation performed in these Jewish populations when required. This information has significance for preventive and personalized medicine.
In conclusion, HFE-HH is not rare in the North African Jewish population and increased recognition before endpoint disease appearance is desirable. The coinheritance of two frequent alleles, H63D and the β-thalassemia trait, among Oriental and North African Jews raises the possibility of genetic interactions contributing to iron overload and needs to be studied further.
REFERENCES
Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399–408.
Mura C, Raguenes O, Ferec C . HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 1999; 93: 2502–2505.
Ostrer HA . Genetic profile of contemporary Jewish populations. Nat Rev Genet 2001; 2: 891–897.
Kedar-Barnes I, Rozen P . The Jewish people: their ethnic history, genetic disorders and specific cancer susceptibility. Fam Cancer 2004; 3: 193–199.
Ben-Sasson HH, Malamat A, Tadmor H . A history of the Jewish people. Cambridge, MA: Harvard University Press, 1976.
Beutler E, Gelbart T . HLA-H mutations in the Ashkenazi Jewish population. Blood Cells Mol Dis 1997; 23: 95–98.
Matas M, Guix P, Castro JA, et al. Prevalence of HFE C282Y and H63D in Jewish populations and clinical implications of H63D homozygosity. Clin Genet 2006; 69: 155–162.
Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ . Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275–278.
Goland S, Kaftouri BN, Shimoni A, Caspi S, Malnick SD . Hemochromatosis mutations are not linked to dilated cardiomyopathy in Israeli patients. Eur J Heart Failure 2004; 6: 547–550.
Miller SA, Dykes DD, Polesky HF . A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Lynas C . A cheaper and more rapid polymerase chain reaction-restriction fragment length polymorphism method for the detection of the HLA-H gene mutations occurring in hereditary hemochromatosis. Blood 1997; 90: 4235–4236.
Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352: 1769–1778.
Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW . A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med 1999; 341: 718–724.
Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J, Robson KJ . Geography of HFE C282Y and H63D mutations. Genet Test 2000; 4: 183–198.
Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med 2008; 358: 221–230.
Ropero P, Briceño O, Mateo M, et al. Frequency of the C282Y and H63D mutations of the hemochromatosis gene (HFE) in a cohort of 1000 neonates in Madrid (Spain). Ann Hematol 2006; 85: 323–326.
Rochette J, Pointon JJ, Fisher CA, et al. Multicentric origin of hemochromatosis gene (HFE) mutations. Am J Hum Genet 1999; 64: 1056–1062.
Cullen LM, Gao X, Easteal S, et al. The hemochromatosis 845 G–>A and 187 C–>G mutations: prevalence in non-Caucasian populations. Am J Hum Genet 1998; 62: 1403–1407.
Asberg A, Hveem K, Thorstensen K, et al. Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol 2001; 36: 1108–1115.
Delatycki MB, Allen KJ, Nisselle AE, et al. Use of community genetic screening to prevent HFE-associated hereditary haemochromatosis. Lancet 2005; 366: 314–316.
Adams PC, Barton JC . Haemochromatosis. Lancet 20071; 370: 1855–1860.
Schmitt B, Golub RM, Green R . Screening primary care patients for hereditary hemochromatosis with transferrin saturation and serum ferritin level: systematic review for the American College of Physicians. Ann Intern Med 2005; 143: 522–536.
Burke W, Imperatore G, McDonnell SM, Baron RC, Khoury MJ . Contribution of different HFE genotypes to iron overload disease: a pooled analysis. Genet Med 2000; 2: 271–277.
Aguilar-Martinez P, Bismuth M, Picot MC, et al. Variable phenotypic presentation of iron overload in H63D homozygotes: are genetic modifiers the cause?. Gut 2001; 48: 836–842.
Beutler E, Felitti V, Gelbart T, Ho N . The effect of HFE genotypes on measurements of iron overload in patients attending a health appraisal clinic. Ann Intern Med 2000; 133: 329–337.
Mura C, Nousbaum JB, Verger P, et al. Phenotype-genotype correlation in haemochromatosis subjects. Hum Genet 1997; 101: 271–276.
Carella M, D'Ambrosio L, Totaro A, et al. Mutation analysis of the HLA-H gene in Italian hemochromatosis patients. Am J Hum Genet 1997; 60: 828–832.
Bukvic N, Sportelli F, Sessa F, et al. Coexistence of beta-thalassemia and hereditary hemochromatosis in homozygosity: a possible synergic effect?. Hemoglobin 2009; 33: 155–157.
Martins R, Picanço I, Fonseca A, Ferreira L, et al. The role of HFE mutations on iron metabolism in beta-thalassemia carriers. J Hum Genet 2004; 49: 651–655.
Yamsri S, Sanchaisuriya K, Fucharoen S, et al. H63D mutation of the hemochromatosis gene and serum ferritin levels in Thai thalassemia carriers. Acta Haematol 2007; 118: 99–105.
Ruiz-Argüelles GJ, Garcés-Eisele J, Reyes-Núñez V, et al. Heterozygosity for the H63D mutation in the hereditary hemochromatosis (HFE) gene may lead into severe iron overload in beta-thalassemia minor: observations in a thalassemic kindred. Rev Invest Clin 2001; 53: 117–120.
Melis MA, Cau M, Deidda F, Barella S, Cao A, Galanello R . H63D mutation in the HFE gene increases iron overload in beta-thalassemia carriers. Haematologica 2002; 87: 242–245.
Filon D, Oron V, Krichevski S, et al. Diversity of β-globin mutations in Israeli ethnic groups reflects recent historic events. Am J Hum Genet 1994; 54: 836–843.
Rund D, Filon D, Doewling CE, Rachmilewitz E, Kazazian HH Jr, Oppenheim A, Diversity of molecular lesions causing beta B-thalassemia in Israel Jewish ethnic groups. In: Adam A, Bonne Tamir B, editors. New perspectives in genetic markers and diseases among the Jewish people. Cambridge, MA: Oxford Press, 1992; 228–236.
Kowdley KV . Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004; 127: S79–S86.
Barton JC, Bertoli LF, Acton RT . HFE C282Y and H63D in adults with malignancies in a community medical oncology practice. BMC Cancer 2004; 4: 6.
Deugnier Y, Turlin B . Iron and hepatocellular carcinoma. J Gastroenterol Hepatol 2001; 16: 491–494.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reish, O., Shefer-Kaufmann, N., Shimshoni, D. et al. Frequencies of C282Y and H63D alleles in the HFE gene among various Jewish ethnic groups in Israel: A change of concept required. Genet Med 12, 122–125 (2010). https://doi.org/10.1097/GIM.0b013e3181cb78d6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181cb78d6
Keywords
This article is cited by
-
Intragenic haplotype analysis of common HFE mutations in the Portuguese population
Journal of Genetics (2015)
