
Abstract
Purpose: To identify a molecular genetic cause in patients with a clinical diagnosis of osteogenesis imperfecta (OI) type I/IV.
Methods: The authors performed multiplex ligation-dependent probe amplification analysis of the COL1A1 gene in a group of 106 index patients.
Results: In four families with mild osteogenesis imperfecta and no other phenotypic abnormalities, a deletion of the complete COL1A1 gene on one allele was detected, a molecular finding that to our knowledge has not been described before, apart from a larger chromosomal deletion detected by fluorescent in situ hybridization encompassing the COL1A1 gene in a patient with mild osteogenesis imperfecta and other phenotypic abnormalities. Microarray analysis in three of the four families showed that it did not concern a founder mutation.
Conclusion: The clinical picture of complete COL1A1 allele deletions is a comparatively mild type of osteogenesis imperfecta. As such, multiplex ligation-dependent probe amplification analysis of the COL1A1 gene is a useful additional approach to defining the mutation in cases of suspected osteogenesis imperfecta type I with no detectable mutation.
Similar content being viewed by others
Main
Osteogenesis imperfecta (OI) constitutes a heterogeneous group of diseases characterized by susceptibility to bone fractures with variable severity and presumed or proven defects in collagen type I biosynthesis.1 The severity of OI ranges from perinatally lethal to occasional fractures.2 The apparent clinical variability in OI has led to the development of the classification by Sillence et al.,3 initially in OI type I (mild, dominantly inherited OI with bone fragility and blue sclerae), II (perinatal lethal), III (progressive deforming), and IV (dominant with normal sclerae and mild deformity). Depending on the age of presentation, OI can be difficult to distinguish from some other genetic and nongenetic causes of fractures, including nonaccidental injury.2,4–6 Recently, rare autosomal recessive causes of lethal and severe OI have been described,7–9 but in the majority of affected individuals, OI is dominantly inherited and caused by a heterozygous mutation in either of the two genes, COL1A1 and COL1A2, encoding the chains of type I collagen. Type I collagen is the major structural protein in bone, tendon, and ligamen. It is first synthesized in the rough endoplasmic reticulum (rER) as type I procollagen, containing C- and N-terminal propeptides. In the rough endoplasmic reticulum, the two alpha-1 chains and the one alpha-2 chain consisting of Gly-X-Y triplets will fold in the C-to-N direction to form a triple helix. During folding, collagen is modified by, among others, specific enzymes that hydroxylate lysine and proline residues and glycosylate hydroxylysyl residues. This process is called posttranslational modification, and it stops as soon as the chain in which the residues are located is folded.10 After folding, the procollagen molecules are transported through the Golgi apparatus in the pericellular environment where cleavage of the N- and C-terminal propeptides occurs and collagen molecules aggregate to form fibrils.11 At present, more than 800 distinct mutations in the COL1A1 and COL1A2 genes have been described to cause OI types II–IV.12 The two mildest forms of OI, OI types I and IV, account for considerably more than half of all OI cases.13 OI types II–IV cases are mostly caused by glycine substitution mutations and splice site mutations, resulting in posttranslational overmodification and synthesis of abnormal collagen type I molecules. In contrast, OI type I is often caused by a nonfunctional COL1A1 allele (null allele)14 because of mutations generating destabilization and rapid degeneration of the mutant COL1A1 mRNA resulting in decreased amount of normal collagen type I molecules.15,16 Both types of abnormalities (abnormal or decreased synthesis of collagen type I) may be detected by electrophoresis of type I collagen synthesized by cultured dermal fibroblasts.17 The presence of normal collagen type I molecules explains the fact that OI type I is the mildest type of OI. OI type I is characterized clinically by increased bone fragility often leading to fractures, ranging from few to 100,13 without secondary deformities in combination with blue sclera, conductive or mixed hearing loss in late adolescence (approximately 50% of cases), not only short but also often normal height, and dentinogenesis imperfecta (approximately 60% of cases). Radiologically, in OI type I, bone fragility in combination with generalized demineralization, slender shafts of tubular bones with thin cortex and poorly trabeculated spongiosa are evident. Furthermore, ossification of the cranial vault is often retarded, leading to a mosaic pattern of Wormian bones.18 In this study, the authors describe four families with OI type I because of a deletion of the complete COL1A1 allele detected by multiplex ligation-dependent probe amplification (MLPA), which to our knowledge has not been described in the literature before.
MATERIALS AND METHODS
Patients
Our DNA laboratory currently has material of 803 index patients with a clinical suspicion of OI; 430 of these index patients have been referred with a clinical diagnosis of OI type I or OI type I/IV. In 100 index patients, a COL1A1 or COL1A2 mutation has been detected. In the remaining 330 index patients, sequence analysis of the COL1A1/2 genes has not yet been completed, or no mutation has been found. From these 330 index patients, we performed MLPA analysis of the COL1A1 gene in 106 patients. In these 106 patients, four index patients with normal COL1A1 and COL1A2 sequencing results were found to have a deletion of the COL1A1 gene. A written informed consent for publication was obtained from these four families according to the journal standard for patient consent. Physical examination was carried out in combination with radiologic surveys. Skin biopsies were available from individuals 1.II.1, 2.I.2, 3.I.2, and 4.I.1. Ninety clinically normal individuals were used as a control group.
Collagen type I electrophoresis
Fibroblast cultures were established under standard conditions. Cells were seeded at 35,000 cells/cm2. Labeling of the fibroblasts and purification of the collagen molecules were performed as reported elsewhere,19 with the exception of performing sodium dodecyl sulfate-electrophoresis without 3% stacking gel. Dried gels were analyzed on a phosphor imager (Molecular Dynamics). Collagen type I electrophoresis was carried out in 39 healthy controls and in affected individuals 1.II.1, 2.I.2, 3.I.2, and 4.I.1.
Genomic DNA analysis
Genomic DNA (gDNA) sequencing of the COL1A1 (NG_007400.1) and COL1A2 (NG_007405.1) genes was performed in individuals 1.II.1, 2.I.2, and 3.I.2. Primer sequences are available on request.
Complementary DNA analysis
RNA was isolated from primary fibroblasts cultured in the presence or absence of cycloheximide (5 mg/mL) using the mini RNA isolation II kit (Baseclear, Leiden, The Netherlands) according to the manufacturer's instructions. To identify the alternatively spliced transcript, complementary DNA (cDNA) was prepared with oligo-dTprimer and Superscript™II RT reverse transcriptase (Invitrogen, San Diego, CA). PCR products were sequenced using a 3730 sequencing system (Applied Biosystems, Foster City, CA). cDNA analysis was performed in individuals 2.I.2, 3.I.2, and 4.I.1.
Multiplex ligation-dependent probe amplification
MLPA analysis was performed as described elsewhere20 with the SALSA MLPA kit P271-B1 COL1A1 that contains 33 probes in the COL1A1 gene of which 26 were used in this study and seven reference probes widely spread over the genome (MRC-Holland, available at: www.mlpa.com).
Microarray
The COL1A1 deletions in Families 2–4 were further characterized by high-resolution microarray using Array CGH. No permission was obtained from Family 1 to perform microarray. Labeling and hybridization procedures were performed according to Buffart et al.21 Briefly, 500 ng gDNA was labeled according to the manufacturer's instructions using the ENZO Genomic DNA Labeling kit (Enzo Life Sciences, Farmingdale, NY). Cy3 label was used for the patient's DNA and Cy5 label for reference DNA (DNA isolated from blood obtained from 18 healthy females). Hybridization of the patient was performed onto 180k arrays, with each array containing approximately 170,000 in situ synthesized 60-mer oligonucleotides, representing 13 KB overall median probe spacing (Agilent Technologies, Palo Alto, CA, genome browser-build hg 18). Images of the arrays were acquired using a microarray scanner G2505B (Agilent Technologies), and image analysis was performed using feature extraction software (version 10.5; Agilent Technologies). The Agilent CGH-v4_91 protocol was applied using default settings. Data analysis was performed with Agilent analysis software (Agilent Genomic Workbench 5.0, Agilent Technologies), with threshold 4.0, algorithm ADM-2 and aberration filter settings for minimum number of probes in Region 2 and minimum absolute average log ratio for Region 0.3.
Marker analysis
Genetic analysis (Powerplex® 16 System, Promega, Madison, WI) was performed to investigate monozygosity in twin sisters II.1 and II.2 from Family 1.
RESULTS
Phenotypical analysis
Family 1
Individual II.1.
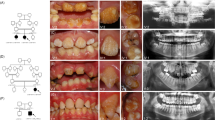
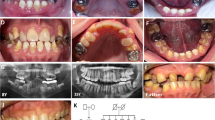
Individual II.1 (Fig. 1) is a 10-year old girl with OI type I. A fracture of the femur was noted shortly after birth. Until now, she has had eight fractures. Her height is 128.5 cm (−2.5 standard deviation [SD]), head circumference is 53.5 cm (0 SD), and bone density of the hips and lumbar vertebral column is between −4 and −7 SD, as measured by dual x-ray absorptometry (DXA). She has Wormian bones, a scoliosis, and gray-blue sclerae typical for OI. She has no dentinogenesis imperfecta.

Pedigrees of Families 1–4. The individuals who have a COL1A1 allele deletion are marked with an asterisk, and the individuals who are clinically affected with OI are depicted in black. Individuals II.1 and II.2 from Family 3 are marked with a question mark because they have no clear diagnosis of OI, and no molecular genetic diagnosis has been performed.
Individual II.2.
Individual II.2 (Fig. 1) is the monozygous twin sister of II.I. She has had no fractures or other signs of OI. Her height is 143.5 cm (−1 SD), and head circumference is 53.2 cm (0 SD).
Family 2
Individual I.2.
Affected individual I.2 (Fig. 1) is a 44-year-old woman. She was diagnosed with OI type I because of multiple fractures in childhood in combination with blue sclerae. She has had 17 fractures in total, with her last fracture occurring at the age of 34 years. She has one-sided hearing loss. At the age of 43 years, she had a cerebrovascular accident that resulted in hemiparesis.
Individual II.1.
The daughter II.1 (Fig. 1) has had two fractures from birth until the age of 5 years. Both fractures occurred after trauma. She has gray-blue sclera but no other features of OI.
Family 3
Individual I.2.
Affected individual I.2 (Fig. 1) is a 37-year-old woman. Her height is 170 cm (0 SD). She has had one fracture at the age of 7 years. Normal bone density measured by DXA was noted. She experiences frequent luxations of her knee and ankle joints. Her face has a mild triangular shape, and her sclerae are blue. She has extensive caries in combination with enamel loss. Her two daughters have not had any fractures, but they have blue sclerae, hypermobility, and enamel loss. No abnormalities on skeletal radiographs of the daughters were noted. Bone density measured by DXA appeared to be reduced in both children. No biochemical or molecular diagnostic testing has been performed in these children.
Family 4
Individual I.1.
Affected individual I.1 (Fig. 1) is a 40-year-old man. His height is 168 cm (0 SD). He has had seven fractures, all occurring before the age of 12 years. His sclerae are blue. He has no hearing loss or dentinogenesis imperfecta. No radiographs are available.
Individual II.1.
Affected individual II.1 is a 1-year-old boy. He has blue sclerae, and has had no fractures.
Electrophoresis of collagen type I
Collagen electrophoresis revealed a reduced but somewhat variable level of type I collagen synthesis. This is expressed in Table 1 by A: the percentage collagen type I of total collagen production (= [collagen alpha-1(I) + collagen alpha-2(I)]/[collagen alpha-1(I) + collagen alpha-2(I) + collagen alpha-1(III)]) and B: the percentage collagen alpha-1(I) of total collagen production (= collagen alpha-1(I)/[collagen alpha-1(I) + collagen alpha-2(I) + collagen alpha-1(III)]. In 39 healthy individuals, A was 91%, and B was 63%, both with an SD of 3%.
Molecular genetic analysis
Molecular genetic analysis with cDNA and gDNA sequencing revealed no mutations in the COL1A1/2 genes.
Multiplex ligation-dependent probe amplification
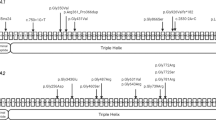
By MLPA, a deletion of the complete COL1A1 allele (Fig. 2, Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A121) was detected in individuals II.1 and II.2 from Family 1; I.2 and II.1 from Family 2; I.2 from Family 3; and I.1 and II.1 from Family 4 (Fig. 1). The average peak ratio of all COL1A1 probes combined in 1.II.2 (0.67) and of 3.I.2 (0.59) may point to mosaicism (Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A121). Complete COL1A1 allele deletions were not detected in 180 chromosomes from clinically normal controls.

MLPA results in individuals from Families 1 to 4, showing a deletion of the complete COL1A1 allele. Twenty-six probes in the COL1A1 gene and seven reference probes are shown. The MLPA COL1A1 probes are listed on the x-axis according to their position in 5′>3′ orientation. The peak ratio is depicted on the y-axis. A heterozygous deletion of the complete COL1A1 allele is visible as an approximately 50% reduced relative peak area of the amplification product of that probe compared with reference probes. The average peak ratio of 0.71 and 0.59 in 1.II.2 and 3.I.2 points to mosaicism, respectively. The peak ratio values can be found in Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A121.
Microarray analysis
Further characterization of the COL1A1 allele deletion using high-resolution microarray revealed that the deletions had different sizes (182–217 kb [Family 2], 512–353 kb [Family 3], and 451–481 kb [Family 4]) containing multiple other genes (Fig. 3), which were studied by using the UCSC browser available at: http://genome.ucsc.edu/.22

Results of microarray analysis. The results of microarray analysis in families 2–4, showing deletions of different sizes indicating that no founder mutation exists.
The deletion in Family 2 encompasses the following reference sequence genes: ITGA3 (NM_002204), PDK2 (NM_002611.3), SAMD14 (NM_174920.2), PPP1R9B (NM_032595.3), SGCA (NM_000023), HILS1 (NR_024193), and COL1A1 (NM_000088.3).
The deletion in Family 3 encompasses the following reference sequence genes: TAC4 (NM_001077504.1), DLX4 (NM_001934), DLX3 (NM_005220) ITGA3, PDK2, SAMD14, PPP1R9B, SGCA, HILS1, COL1A1, TMEM92 (NM_153229.2), XYLT2 (NM_022167.2), and MRPL27 (NM_016504.2).
The deletion in Family 4 encompasses the following reference sequence genes: DLX4, DLX3, ITGA3, PDK2, SAMD14, PPP1R9B, SGCA, HILS1, COL1A1, TMEM92, XYLT2, MRPL27, EME1 (NM_001166131.1), and LRRC59 (NM_018509.2).
The DLX3, SGCA, and XYLT2 genes are disease-associated genes apart from COL1A1.
Marker analysis
Genetic analysis (concordance in 16 short tandem repeat markers) confirmed monozygosity of 1.II.1 and 1.II.2 (Powerplex® 16 System, Promega, Madison, WI).
DISCUSSION
We describe five families in which individuals are affected with OI because of a deletion of a complete COL1A1 allele. Most affected individuals in our four families have a clinical phenotype consistent with OI type I. Interestingly, the affected individual II.2 from Family 1 seems to have no symptoms of OI at all, whereas her monozygous twin sister with the same deletion has a phenotype fitting the diagnosis OI type I. No deletion was found in DNA from blood samples of the parents. The most likely explanation is that the twin sisters are mosaic with a postzygotic mutation leading to unequal grades of somatic mosaicism. The average peak ratio of the COL1A1 MLPA probes in 1.II.2 (0.67) may point to mosaicism in leukocytes. Unfortunately, we were not able to obtain cell samples from different tissues in both twin sisters to further investigate this. In Family 1, the clinical variability in individuals with a COL1A1 deletion may be due to mosaicism for the COL1A1 deletion.
OI type I almost always results from mutations in one COL1A1 allele that introduce a premature termination codon and decrease the stability of mRNA. These mutations may consist of nucleotide substitutions, frame shifts, and splice site mutations. Large deletions of the collagen type I genes are infrequent in OI.23 Interestingly, the first mutation described to cause OI was a multiexon COL1A1 deletion eliminating exons 23–25 resulting in OI type II.24 In 1996, another multiexon deletion was published, namely a 562-bp exon to intron deletion extending from the last codon of exon 34 to intron 36.25 A genomic deletion of exons 40–48 was recently reported, causing OI type II.26 In the COL1A2 gene, three multiexon deletions have been described to cause OI type II27 and OI type IB.23 So, it is generally accepted that in-frame partial deletions in the COL1A1 or COL1A2 genes can result in a lethal or severe OI phenotype when the protein is not rapidly degraded but instead when it is incorporated in the triple helix exerting a dominant negative effect. Interestingly, a 17q21.33 to q23.1 deletion including the whole COL1A1 gene was described to cause a mild OI phenotype with blue sclerae, osteopenia, and a fracture at birth in combination with micrognathia, cleft palate, and marfanoid features.28 We state that deletions of the complete COL1A1 allele do not have the above-described dominant negative effect on COL1A1 expression and thereby result in a milder clinical picture. The clinical phenotype in most of our patients with a complete COL1A1 deletion is comparable with the phenotype of OI type I seen in patients with a nonfunctional COL1A1 allele. Microarray analysis revealed that no common deletion of COL1A1 exists in our families. In Families 3 and 4, the DLX3 gene was also deleted. A 4-bp deletion in the DLX3 gene has been described to cause trichodentoosseous syndrome (Online Mendelian Inheritance in Man #190320) characterized by kinky or curly hair, dolichocephaly enamel hypoplasia, increased dental caries, radial dense bones, and occasionally brittle nails. The observed mutation was predicted to cause a frame shift and premature termination codon, resulting in a functionally altered DLX3 protein with a potential dominant negative effect. A 2-bp deletion in the DLX3 gene seemed to be responsible for the characteristic dental phenotype without hair or bone abnormalities.29 Interestingly, Patient I.2 from Family 3 has extensive caries in combination with enamel loss, which is a feature of OI but has also been described in patients with a deletion in the DLX3 gene. However, Patient I.1 from Family 4 has no teeth abnormalities. Therefore, it cannot be concluded that a deletion of the DLX3 gene affected the clinical phenotype in our patients. Given the function, when known, of the other deleted reference sequence genes, it is unlikely that they contribute to the phenotype of the patients of Families 2–4 described in this article.
Sequence analysis of COL1A1 and COL1A2 cDNA, which detects mutations in the coding sequence, and of gDNA, which detects mutations that either alter the sequence or the stability of mRNA, identifies mutations in approximately 90% of patients with OI type I. Measuring the amount and structure of type I procollagen molecules synthesized by dermal fibroblasts also detects approximately 90% of individuals known to have OI.2,13 It is likely that the combination of biochemical and molecular genetic analysis would identify a small number of additional individuals with OI.4 Indeed, the patients we describe with deletions of a complete COL1A1 gene would not have been diagnosed with OI if only cDNA and/or gDNA sequencing would have been performed. As such, in cases of suspected OI type I without a detectable mutation, MLPA analysis of the COL1A1 gene is a useful additional approach to defining the mutation. This has important implications for the prenatal diagnostic approach, before a pregnancy, in patients with suspected OI type I and reduced type I procollagen synthesis, because the reduced synthesis of type I collagen chains cannot be detected reliably in chorionic villi cells.2 Interestingly, in our families, three patients (2.I.2, 3.I.2, and 4.I.1) had minimally reduced amount of collagen type I, making the diagnosis of OI purely based on biochemical testing unreliable. The observation that minimally diminished collagen type I production, as measured in fibroblasts, still causes OI type I might be due to tissue-specific expression of collagen type I with the regulation of expression in skin, or dermal fibroblasts, and bone30 being different.
In conclusion, in patients with suspected OI type I, performing collagen type I electrophoresis and sequencing of the COL1A1/2 genes are insufficient to rule out OI type I when only gDNA is available and no heterozygous polymorphic variants are detected. In view of the clinical and biochemical characteristics of complete COL1A1 allele deletions, MLPA should be performed as well, even in the presence of near-normal or normal collagen type I production, as determined by collagen type I electrophoresis.
References
van Dijk FS, Pals G, Rijn van RR, Nikkels PGJ, Cobben JM . Classification of osteogenesis imperfecta revisited. Eur J Med Genet 2010; 53: 1–5.
Byers PH, Krakow D, Nunes ME, Pepin M . Genetic evaluation of suspected osteogenesis imperfecta. Genet Med 2006; 8: 383–388.
Sillence DO, Senn A, Danks DM . Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16: 101–116.
Marlowe A, Pepin MG, Byers PH . Testing for osteogenesis imperfecta in cases of suspected non-accidental injury. J Med Gen 2002; 39: 282–286.
Ablin DS, Greenspan A, Reinhart M, Grix A . 1990. Differentiation of child abuse from osteogenesis imperfecta. AJR 1990; 154: 1035–1046.
Steiner RD, Pepin M, Beyers PH . Studies of collagen synthesis and structure in the differentiation of child abuse from osteogenesis imperfecta. J Pediatr 1996; 128: 542–547.
Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 2006; 355: 2757–2764.
Cabral WA, Chang W, Barnes AM, et al. Prolyl 3-hyrdroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007; 39: 359–365.
van Dijk FS, Nesbitt IM, Zwikstra EH, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009; 85: 521–527.
Engel J, Prockop DJ . The zipper-like folding of collagen triple helices and the effects of mutations that might disrupt the zipper. Annu Rev 1991; 20: 137–152.
Byers PH, Wallis GA, Willing MC . Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet 1991; 28: 433–442.
Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007; 28: 209–221.
Steiner RD, Pepin MG, Byers PH Osteogenesis imperfecta, 2005. Available at: http://www.genetests.org. Accessed April 4, 2010.
Willing MC, Pruchno CJ, Atkinson M, Byers PH . Osteogenesis imperfecta type I is commonly due to a COL1A1 null allele of Type I collagen. Am J Hum Genet 1992; 51: 508–515.
Willing MC, Deschenes SP, Scott DA, et al. Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen. Am J Hum Genet 1994; 55: 638–647.
Nuytinck L, Sayli BS, Wettinck K, De Paepe A . Prenatal diagnosis of osteogenesis imperfecta type I by COL1A1 null-allele testing. Prenat Diagn 1999; 19: 873–875.
Wenstrup RJ, Willing MC, Starman BJ, Byers PH . Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am J Hum Genet 1990; 46: 975–982.
Spranger J, Brill P, Poznanski A Bone dysplasias. An atlas of genetic disorders of skeletal development. In: Spranger J, Brill P, Poznanski A (eds) Section X: skeletal dysplasias with prominent diaphyseal involvement, osteogenesis imperfecta, type III/IIB. Oxford, Oxford University Press, 2003; 440–441.
Körkkö J, Ala-Kokko L, De Paepe A, Nuytinck L, Earley J, Prockop DJ . Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfect type I: identification of common sequences of null-allele mutations. Am J Hum Genet 1998; 62: 98–110.
Schouten JP, McWlgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G . Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucl Acids Res 2002; 30: e57.
Buffart TE, Israeli D, Tijssen M, et al. Across arraycomparative genomic hybridization: a strategy to reduce reference channel hybridizations. Genes Chromosomes Cancer 2008; 47: 994–1004.
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
Mundlos S, Chan D, Weng YM, Sillence DO, Cole WG, Bateman JF . Multiexon deletions in the type I collagen COL1A2 gene in osteogenesis imperfecta type 1B. J Biol Chem 1996; 271: 21068–21074.
Barsh GS, Roush CL, Bonadio J, Byers PH, Gelinas RE . Intron-mediated recombination may cause a deletion in an alpha1 type I collagen chain in a lethal form of osteogenesis imperfecta. Proc Natl Acad Sci USA 1985; 82: 2870–2874.
Wang O, Forlino A, Marini JC . Alternative splicing in COL1A1 mRNA leads to a partial null allele and two in-frame forms with structural defects in non-lethal osteogenesis imperfecta. J Biol Chem 1996; 271: 28617–28623.
Bodian DL, Chan T, Poon A, et al. Mutation and polymorphism spectrum in osteogenesis imperfecta type II. Implications for genotype-phenotype relationships. Hum Mol Genet 2009; 18: 463–471.
Willing MC, Cohn DH, Starman B, Holbrook KA, Greenberg CR, Byers PH . Heterozygosity for a large deletion in the alpha2(I) collagen gene has a dramatic effect on type I collagen secretion and produces perinatal lethal osteogenesis imperfecta. J Biol Chem 1988; 263: 8398–8404.
Pollitt R, McMahon R, Nunn J, et al. Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum Mutat 2006; 27: 716.
OMIM (Online Mendelian Inheritance in man). Baltimore: John Hopkins University, Center for Medical Genetics, 1996 Available at: http://www3.ncbi.nlm.nih.gov/omim/(September). Accessed April 4, 2010.
Lichtler A, Stover M, Angilly J, Kream B, Rowe DW . Isolation and characterization of the rat alpha (I) collagen promoter. J Biol Chem 1989; 264: 3072–3077.
Acknowledgements
The authors thank Isabel M. Nesbitt from the Sheffield Molecular Genetics Service, Sheffield Children's National Health Service Foundation Trust, Sheffield Children's Hospital, United Kingdom, for her technical assistance and the patients for their kind cooperation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
van Dijk, F., Huizer, M., Kariminejad, A. et al. Complete COL1A1 allele deletions in osteogenesis imperfecta. Genet Med 12, 736–741 (2010). https://doi.org/10.1097/GIM.0b013e3181f01617
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181f01617
Keywords
This article is cited by
-
Tantalum implanted entangled porous titanium promotes surface osseointegration and bone ingrowth
Scientific Reports (2016)
-
Osteogenesis Imperfecta Type I Caused by COL1A1 Deletions
Calcified Tissue International (2016)
-
EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta
European Journal of Human Genetics (2012)
-
No evidence for disturbed COL1A1 and A2 expression in otosclerosis
European Archives of Oto-Rhino-Laryngology (2012)
