
Abstract
Purpose: Accumulation of mitochondrial DNA deletions and the resultant impaired oxidative phosphorylation may play a pathogenic role in the mediation of age-related sarcopenia.
Methods: Twenty four participants of the New Mexico Aging Process Study were classified as normal lean (n = 15) or sarcopenic (n = 9) based on body composition determined by Dual Energy x-ray Absorptiometry. Complex I and Complex IV activities were measured in the skeletal muscle samples obtained from gastrocnemius muscle. A two-stage nested polymerase chain reaction strategy was used to identify the mitochondrial DNA deletions in the entire mitochondrial genome in the skeletal muscle samples.
Results: Although Complex I activity was not significantly different (5.5 ± 0.9 vs. 4.6 ± 0.7 mU/mg protein, P > 0.05), Complex IV activity was higher in sarcopenic subjects (1.4 ± 0.3 vs. 1.0 ± 0.1 mU/mg protein, P < 0.05). Mitochondrial DNA deletions were mostly located in the region of Complex I and spanned from nicotinamide adenine dinucleotide dehydrogenase 1 to nicotinamide adenine dinucleotide dehydrogenase 6. Deletions in the 8,577–10,407 bp and 10,233–11,249 bp regions were associated with a significant decrease in Complex I activity (P < 0.05 and P = 0.02, respectively). Total cumulative deletion, defined as the sum of individual length of deletions in a subject, was comparable in subjects with and without sarcopenia (1760 ± 726 vs. 1782 ± 888 bp, P > 0.05). The magnitude of mitochondrial DNA deletion, however, correlated positively with lean body mass (r = 0.43, P < 0.05).
Conclusion: Thus, mitochondrial DNA deletions are common in elderly subjects and are negatively related to Complex I activity. The positive association between mitochondrial DNA deletions and lean body mass needs to be confirmed by studies in a larger study population.
Similar content being viewed by others


Main
Skeletal muscle atrophy, or “sarcopenia,” is highly prevalent in the aging population. The prevalence of sarcopenia increases from about 8 to 15% in those 60–80 years of age, to as high as 40% in those older than 80 years.1 Elderly men and women with low relative skeletal muscle mass have higher risk of disability, and two to three times the risk of falls, compared with those with normal muscle mass.2 Multiple factors contribute to the development and progression of sarcopenia, including neuromuscular3 and hormonal4 changes, malnutrition,5 chronic inflammation,6 and reduced physical activity.7 The cellular mechanisms underlying sarcopenia remain an area of active research. Age-related decreases in the amount of functionally intact mitochondria with concomitant decline in cellular production of adenosine triphosphate (ATP), energy-dependent protein synthesis, and increased peroxide leakage may contribute to sarcopenia.8 All mitochondrial-encoded polypeptides are components of the electron transport—oxidative phosphorylation system. Therefore, mutations of the mitochondrial genome are most likely to affect these critical energy transduction pathways. Mitochondrial DNA (mtDNA) is more susceptible to oxidative attack because of its proximity to the respiratory chain in the inner mitochondrial membrane, lack of protective histone-like proteins, and its poor repair activity against damage.9 Deletions and point mutations in mtDNA have been found to correlate with functional decline in the mitochondrial oxidative phosphorylation capacity in muscles with aging.10,11
We hypothesized that the prevalence and severity of mtDNA deletions would be increased in sarcopenic subjects resulting in reduced activity of the electron transport enzyme activity. Results from this study indicate that deletions are predominantly located in the Complex I region and are negatively related to mitochondrial Complex I activity and positively with lean body mass.
MATERIALS AND METHODS
Subjects
Our present study was approved by the University of New Mexico Institutional Review Board and we have obtained informed consent from all participants. Study participants were recruited from the participants of the New Mexico Aging Process Study. Subjects with myopathies, severe peripheral arterial disease, or cancer cachexia, renal disease, and those who were taking corticosteroids, anabolic hormones, or any other medications that might affect muscle mass and function were excluded. Participants were admitted to the General Clinical Research Center at the University of New Mexico the day before the experiment and were maintained on restricted mobility and standard diet.
Dual energy x-ray absorptiometry
A Lunar DPX™ dual energy x-ray absorptiometer (Lunar Radiation Corp., Madison, WI) was used to measure; total soft-tissue mass, % body fat, lean soft-tissue mass, and appendicular skeletal muscle mass. Percent coefficients of variation (%CV) are 1.15% for total body fat mass, 0.51% for total body lean mass, 2.01% for lower extremity fat mass, 1.3% for lower extremity lean mass, 2.96% for upper extremity fat mass, and 1.96% for upper extremity lean mass.
Defining sarcopenia
Sarcopenia was defined as described by Baumgartner et al.12 Because muscle mass is correlated strongly with skeletal size, an index of sarcopenia that expresses muscle mass was adjusted for variation in size.12 Appendicular skeletal muscle (ASM) mass was divided by stature-squared (ASM/Ht2) to provide an index of relative muscle mass. Using this criteria, the subjects were classified as normal lean (normal fat and lean body mass) with an ASM index ≥5.7 and body fat <41% for female, ASM index >7.6, and body fat <30.6% for male. Sarcopenic subjects were classified by normal fat and low lean body mass with an ASM index ≤5.7 and body fat <41% for female, ASM index ≤7.6 and body fat <30.6% for male.
Physical activity assessment
Physical activity was assessed in all subjects using a modified interview administered questionnaire developed and validated by Voorrips et al.13 Household and exercise scores were summed to achieve a total activity score. All subjects reported consistent levels of physical activity for the year before the study.
Muscle biopsy
Biopsies were taken from the medial portion of gastrocnemius using a Bergstrom biopsy needle as described earlier.14 Briefly, the needle was advanced through a small incision and about 30–60 mg of muscle tissue was taken. Fat and connective tissue were removed, and the sample was immediately frozen in liquid nitrogen and stored at −80°C for future analysis.
Analysis of mtDNA deletions
Preparation of DNA from skeletal muscle
DNA was isolated from approximately 5 mg of muscle biopsies using the protocol from commercially available kit Qiagen Micro QIAamp® DNA (Qiagen Inc., Valencia, CA). Briefly, the muscle biopsies were homogenized using repeated freeze and thaw under liquid nitrogen and resuspended in a buffer containing 10 mM Tris-HCL, 10 mM NaCl, and 25 μM EDTA. Using the Qiagen kit, we digested the muscle and isolated total DNA free of protein and RNA. DNA was resuspended in deionized water and quantified using the reading at 260/280 Å.
Amplification of the mitochondrial genome using two-stage nested PCR
mtDNA content in the total genomic DNA isolated from whole tissue was at very low abundance. To improve the specificity for mtDNA in the background of nuclear DNA, a repetitive two-stage nested polymerase chain reaction (PCR) strategy was used to improve the DNA product yield and specificity significantly.15 The outer most primers of mtDNA (Table 1) were designed to amplify the mtDNA from the total genomic DNA using the standard PCR conditions for 25 cycles. In the second repeated nested PCR reaction, primers binding to internal sites in the target first-reaction-product were used to obtain a very distinct PCR product. The PCR primers were designed using the primer-3 program at http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi. Thirteen PCR reactions were performed using primers binding to internal sites in the second-reaction-product to amplify the mtDNA across the whole mitochondrial genome. PCR products were then visualized and sized by agarose gel electrophoresis.
Restriction digests
Restriction fragment length polymorphism protocol was used to further map the smaller sites in mtDNA as part of scanning the whole mitochondrial genome. The DNA used in this procedure was the mtDNA amplified in the second reaction PCR fragments. The restriction enzymes were designed to create smaller fragments specific for the mtDNA position and were used in the restriction fragment length polymorphism protocol (Table 2). Unpurified PCR product (2.5 μL) was digested for 16 hours at 37°C, followed by enzyme deactivation for 10 minutes at 65°C using per 5 μL reaction, 0.5 μL restriction enzyme, 0.5 μL respective buffer (plus BSA if required), and 2.5 μL respective PCR product x μL H2O to 5 μL. Digested products were mixed with 5 μL 20 mM EDTA for a final concentration of 10 mM before loading on to the DNA 1000 lab chips. One-microliter aliquots were analyzed on the Agilent 2100 Bioanalyzer per manufacturer's instructions.
Mitochondrial enzyme activity
Complex I
The method for determining mitochondrial Complex I activity was modified from a protocol developed by Wiedemann et al.16 The rotenone-sensitive portion was subtracted from the total activity and the Complex I enzyme activity (milliunits/mg protein) was calculated by dividing the change in absorbance per minute over the extinction coefficient of nicotinamide adenine dinucleotide (NADH) (5.5 mm–1 cm−1) and protein concentration of each homogenate determined by the Lowry protein assay method.
Complex IV
The method for determining mitochondrial Complex IV activity was based upon a protocol developed by Hess and Pope17 and modified by Hevner et al.18 and Gonzales-Lima and Jones.19 Change in absorbance per minute was standardized to the extinction coefficient of (reduced − oxidized) cytochrome c (19.6 mM−1 · cm−1) and protein concentration of each homogenate determined by the Lowry protein assay method.
Statistical methods
Statistical analyses were conducted using SAS software (SAS, version 9.1, Cary, NC), and α was set at <0.05 for all tests. Total cumulative deletion was defined as the sum of individual deletions in a subject. Data are expressed as mean and SEM. Student t test and Fisher exact test were used to analyze continuous and categorical variables, respectively. The relationship between variables was determined from linear regression analysis and Pearson correlation coefficient. To test whether presence or magnitude of mtDNA deletion modulates Complex I and Complex IV activities, we performed stepwise regression analysis and verified the findings with “best subset” regression. The same procedure was repeated to identify the association with ASM index.
RESULTS
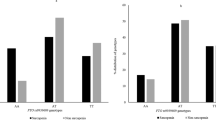
Among the 24 participants, nine had evidence of sarcopenia. Patient characteristics are described in Table 3. Although Complex I activity was comparable, Complex IV activity was significantly higher in sarcopenic participants, compared with normal lean subjects (P < 0.05). The frequency and distribution of mtDNA deletions in the entire study participants is shown in Figure 1. Deletions were mostly located in the region of Complex I and spanned from NADH dehydrogenase (ND) 1 to ND6 (Table 4). mtDNA deletions at one or more sites were observed in 66.7% of subjects with sarcopenia and 77.8% of normal lean participants. The total cumulative deletion was comparable in the two groups (1760 ± 726 vs. 1782 ± 888 bp, P > 0.05). The 12,175–13,708 bp deletion was, however, observed only in subjects with sarcopenia (P = 0.04). Total cumulative deletion correlated positively with lean body mass (r = 0.43, P < 0.05) (Fig. 2). Presence of mtDNA deletion was associated with decreased Complex I activity. Specifically, deletions in the 8,577–10,407 bp and 10,233–11,249 bp regions were associated with significant decrease in Complex I activity (P < 0.05 and P = 0.02, respectively). Activity score (189.3 ± 7.4 vs. 277.2 ± 25.5 units, P < 0.05) was significantly lower in participants with 12,175–13,708 bp deletions. Multivariate analysis did not reveal that any combination of deletions from different sites predicted Complex I or Complex IV activity better than that identified by univariate analysis. Stepwise regression did not show any significant associations between mtDNA deletions and enzyme activity or body composition.

Correlation between total cumulative mtDNA deletion and lean body mass (LBM).

Distribution of mitochondrial DNA deletions among the study participants including both normal lean and sarcopenic subjects.
DISCUSSION
Human mtDNA is a 16,569 bp double-stranded closed circular DNA molecule which codes for 13 polypeptides that are essential for oxidative phosphorylation, 22 tRNAs, and two rRNAs required for mitochondrial protein synthesis.20 The number of mitochondria and also the mtDNA copy number per mitochondrion is organ specific and changes in response to the energy demands of the cell.21 According to the “mitochondrial-lysosomal axis theory of aging,” mitochondria and lysosomes of postmitotic cells undergo profound regressive changes with age, ultimately leading to accumulation of dysfunctional mitochondria and ineffective removal of dysfunctional cellular elements that are responsible for reduced energy availability impaired cellular function and eventually cell death.22 Human studies suggest an increase in and a correlation between mtDNA mutations and the occurrence of skeletal muscle abnormalities with advancing age.23 The 4977 bp deletion of mtDNA, which removes a DNA segment from the nucleotide position 8,483–13,459 has been established as the most common and abundant large scale deletion of mtDNA in various human tissues.24,25 Unlike most published work, we did not specifically target a specific site of mtDNA but walked through the entire genome to seek the association between mtDNA deletion and sarcopenia of aging. Results from this study show that deletions in the Complex I region of the mtDNA are common in elderly subjects and are associated with decrease in Complex I enzyme activity.
In human mitochondria, the 4977 bp deletion accumulates with age in several tissues, including skeletal muscle. The deletion level is determined by mitotic activity, cell size, and metabolic activity of the cell.25 Based on the observation that direct repeat sequence are present at the deletion break points, slipped-strand mispairing mechanism is proposed as a means by which deletions are produced.26 In human mitochondria, the 4977 bp deletion is often flanked by a 13 bp direct repeat.25 Although we did not observe the signature mtDNA4977 deletion, but we did observe deletions spanning across this region (8,577–10,407 bp and 10,866–12,420 bp), which covers the region of the common 4977 bp deletion. The magnitude of mtDNA deletions was not increased in sarcopenic subjects compared with normal lean participants. Contrary to the expectation, the total cumulative deletion correlated positively with lean body mass. This apparent discrepancy in finding could be in the latter analysis where we used ASM index as a continuous variable, which improves the sensitivity and potential for identifying association. Subjects with sarcopenia had a unique deletion in the mitochondrially encoded ND5 region (12,175–13,708 bp), which is a hotspot for mtDNA deletions and is associated with disease states27,28 and aging.29 Interestingly, aged human muscles are reported to have a higher mtDNA/nuclear DNA ratio than young, suggesting that cells may attempt to compensate for an inefficient ATP production per mitochondrion by up-regulating respiratory enzymes.11,30 The mitochondrial genome undergoes continuous turnover with dynamic replication and replacement with a half-life of a few weeks.31 However, damaged and mutated mtDNA molecules continue to be generated faster in disease states resulting in accumulation of abnormal mtDNA.32 Inability to generate new functional mtDNA eventually leads to energy depletion, impaired cellular functions, and eventually to apoptosis of muscle cells. de Grey33 proposed that damaged mitochondria are degraded more slowly than intact mitochondria and that abnormal mitochondria accumulate in the cell by survival of the slowest. The low proton gradient in these inert mitochondria may lead to actually decreased generation of oxygen free radicals, which is implicated in muscle wasting.34,35 However, some studies examining the effect of blocking ATP synthesis in cultured skeletal muscle in vitro have failed to demonstrate any protein wasting effect, either by blocking anaerobic ATP synthesis36 or by blocking aerobic (mitochondrial) ATP synthesis.37 Although classified as sarcopenic, the self-reported activity level in normal lean and sarcopenic participants was comparable. Regular exercise is a potent and effective countermeasure for skeletal muscle aging, possibly through mitochondrial regeneration.38,39 However, exercise capacity may progressively deteriorate with accumulation of mtDNA deletions. We observed that activity level is decreased in subjects with deletions in ND5 region.
Mutations affecting mitochondrion-encoded subunits are also known to be responsible for many hereditary diseases in humans.40 However, because of the heteroplasmic state of the cells that contain both normal and mutant mitochondria and to the presence of allelic variants, it is often difficult to establish the real impact of these mutations on Complex I assembly and activity in patients. In this respect, we observed deletions in the 8,577–10,407 bp and 10,233–11,249 bp regions associated with significant decrease in Complex I activity as has been reported by Hinokio et al.41 They reported 5778-bp deletion (8,214–13,991) in mtDNA from the muscle and liver biopsy specimens linked to the defect in Complex I activity. They further suggested that 5778-bp deletion might cause a defect in mitochondrial oxidative phosphorylation and contribute to the pathogenesis of diabetic amyotrophy and myoatrophy with diabetic nephropathy. The reported deletion covers areas coding ND3, ND4, ND4L, and ND5 in Complex I.
Oxidative phosphorylation is a process in which electrons are passed along a series of carrier molecules called the electron transport chain. The electron transport chain consists of four respiratory enzyme complexes: (a) NADH, ubiquinone oxidoreductase; (b) succinate, ubiquinone oxidoreductase; (c) ubiquinol, cytochrome c oxidoreductase; and (d) cytochrome c oxidase, coupled to ATP synthase.42 We opted to measure Complex I activity because 7 of 13 structural genes in mtDNA encode for polypeptides in Complex I, and hence, it is most likely to be affected by mtDNA deletions.43 Unsurprisingly, we observed a correlation between mtDNA deletion and Complex I activity. Investigators have shown that fibers with electron transport system abnormalities had large mtDNA deletions.10 Deficiency of cytochrome c oxidase is observed in muscle fibers exhibiting sarcopenia44 The reported mitochondrial enzyme activities vary with the animal model, method used, and also the organ studied.43,45 The results of mitochondrial Complex I and Complex IV enzyme activity is consistent with that reported in the literature.16 We noted that the Complex IV activity was higher in sarcopenic subjects. It is unclear if increased enzyme activity was due to a compensatory, but temporary, increase in enzyme activity which precedes the final and irreversible decline. On the other hand, the measured activities may not necessarily reflect the activities in the intact mitochondria because the mitochondria in the assayed homogenates from the muscle biopsies are predominantly in the form of broken membranes so that the measured activities are no longer regulated by the tight coupling of electron transport to oxidative phosphorylation that is usually observed in intact mitochondria. Increased production of reactive oxygen species which occurs as a bi-product of electron transport is a well-established trigger for protein wasting.37
To summarize, we observed that the prevalence of mtDNA deletions were common in older individuals and was predominantly confined to the coding region of Complex I. Total cumulative mtDNA deletion correlated negatively with Complex I activity but positively with lean body mass. The positive association between mtDNA deletions and lean body mass could be due to compensatory replication of mtDNA with resultant accumulation of abnormal mtDNA or mitochondria-related assembly of oxidative stress. These results need to be confirmed by studies in a larger study population.
REFERENCES
Baumgartner RN . Body composition in healthy aging. Ann N Y Acad Sci 2000; 904: 437–448.
Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ . Predictors of skeletal muscle mass in elderly men and women. Mech Ageing Dev 1999; 107: 123–136.
Vandervoort AA . Aging of the human neuromuscular system. Muscle Nerve 2002; 25: 17–25.
Iannuzzi-Sucich M, Prestwood KM, Kenny AM . Prevalence of sarcopenia and predictors of skeletal muscle mass in healthy, older men and women. J Gerontol A Biol Sci Med Sci 2002; 57: M772–M777.
Dreyer HC, Volpi E . Role of protein and amino acids in the pathophysiology and treatment of sarcopenia. J Am Coll Nutr 2005; 24: 140S–145S.
Visser M, Pahor M, Taaffe DR, et al. Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J Gerontol A Biol Sci Med Sci 2002; 57: M326–M332.
Castillo EM, Goodman-Gruen D, Kritz-Silverstein D, Morton DJ, Wingard DL, Barrett-Connor E . Sarcopenia in elderly men and women: the Rancho Bernardo study. Am J Prev Med 2003; 25: 226–231.
Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA 2005; 102: 5618–5623.
Clayton DA, Doda JN, Friedberg EC . The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci USA 1974; 71: 2777–2781.
Cao Z, Wanagat J, McKiernan SH, Aiken JM . Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser-capture microdissection. Nucleic Acids Res 2001; 29: 4502–4508.
Pesce V, Cormio A, Fracasso F, et al. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic Biol Med 2001; 30: 1223–1233.
Baumgartner RN, Koehler KM, Gallagher D, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147: 755–763.
Voorrips LE, Meijers JH, Sol P, Seidell JC, van Staveren WA . History of body weight and physical activity of elderly women differing in current physical activity. Int J Obes Relat Metab Disord 1992; 16: 199–205.
Raj DS, Moseley P, Dominic EA, et al. Interleukin-6 modulates hepatic and muscle protein synthesis during hemodialysis. Kidney Int 2008; 73: 1054–1061.
van Den Bosch BJ, de Coo RF, Scholte HR, et al. Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Res 2000; 28: E89.
Wiedemann FR, Vielhaber S, Schroder R, Elger CE, Kunz WS . Evaluation of methods for the determination of mitochondrial respiratory chain enzyme activities in human skeletal muscle samples. Anal Biochem 2000; 279: 55–60.
Hess HH, POPE A . Ultramicrospectrophotometric determination of cytochrome oxidase for quantitative histochemistry. J Biol Chem 1953; 204: 295–306.
Hevner RF, Liu S, Wong-Riley MT . An optimized method for determining cytochrome oxidase activity in brain tissue homogenates. J Neurosci Methods 1993; 50: 309–319.
Gonzalez-Lima F, Jones D . Quantitative mapping of cytochrome oxidase activity in the central auditory system of the gerbil: a study with calibrated activity standards and metal-intensified histochemistry. Brain Res 1994; 660: 34–49.
Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature 1981; 290: 457–465.
Veltri KL, Espiritu M, Singh G . Distinct genomic copy number in mitochondria of different mammalian organs. J Cell Physiol 1990; 143: 160–164.
Brunk UT, Terman A . The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 2002; 269: 1996–2002.
Cormio A, Milella F, Vecchiet J, Felzani G, Gadaleta MN, Cantatore P . Mitochondrial DNA mutations in RRF of healthy subjects of different age. Neurobiol Aging 2005; 26: 655–664.
Wei YH . Oxidative stress and mitochondrial DNA mutations in human aging. Proc Soc Exp Biol Med 1998; 217: 53–63.
Cortopassi GA, Shibata D, Soong NW, Arnheim N . A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA 1992; 89: 7370–7374.
Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace DC . Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: a slip-replication model and metabolic therapy. Proc Natl Acad Sci USA 1989; 86: 7952–7956.
Nishigaki Y, Marti R, Hirano M . ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum Mol Genet 2004; 13: 91–101.
Tanaka M, Sato W, Ohno K, Yamamoto T, Ozawa T . Direct sequencing of deleted mitochondrial DNA in myopathic patients. Biochem Biophys Res Commun 1989; 164: 156–163.
Torii K, Sugiyama S, Tanaka M, et al. Aging-associated deletions of human diaphragmatic mitochondrial DNA. Am J Respir Cell Mol Biol 1992; 6: 543–549.
Lezza AM, Pesce V, Cormio A, et al. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. FEBS Lett 2001; 501: 74–78.
Menzies RA, Gold PH . The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 1971; 246: 2425–2429.
Kopsidas G, Kovalenko SA, Heffernan DR, et al. Tissue mitochondrial DNA changes. A stochastic system. Ann N Y Acad Sci 2000; 908: 226–243.
de Grey AD . A proposed refinement of the mitochondrial free radical theory of aging. Bioessays 1997; 19: 161–166.
Kowald A . The mitochondrial theory of aging: do damaged mitochondria accumulate by delayed degradation?. Exp Gerontol 1999; 34: 605–612.
Grune T, Merker K, Sandig G, Davies KJ . Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun 2003; 305: 709–718.
Bevington A, Brown J, Pratt A, Messer J, Walls J . Impaired glycolysis and protein catabolism induced by acid in L6 rat muscle cells. Eur J Clin Invest 1998; 28: 908–917.
Bevington A, Brown J, Walls J . Leucine suppresses acid-induced protein wasting in L6 rat muscle cells. Eur J Clin Invest 2001; 31: 497–503.
Johnston AP, De LM, Parise G . Resistance training, sarcopenia, and the mitochondrial theory of aging. Appl Physiol Nutr Metab 2008; 33: 191–199.
Taivassalo T, Haller RG . Implications of exercise training in mtDNA defects—use it or lose it?. Biochim Biophys Acta 2004; 1659: 221–231.
Crimi M, Sciacco M, Galbiati S, et al. A collection of 33 novel human mtDNA homoplasmic variants. Hum Mutat 2002; 20: 409.
Hinokio Y, Suzuki S, Komatu K, et al. A new mitochondrial DNA deletion associated with diabetic amyotrophy, diabetic myoatrophy and diabetic fatty liver. Muscle Nerve 1995; 3: S142–S149.
Saraste M . Structural features of cytochrome oxidase. Q Rev Biophys 1990; 23: 331–366.
Lenaz G, Bovina C, Castelluccio C, et al. Mitochondrial complex I defects in aging. Mol Cell Biochem 1997; 174: 329–333.
Bua EA, McKiernan SH, Wanagat J, McKenzie D, Aiken JM . Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J Appl Physiol 2002; 92: 2617–2624.
Pitkanen S . Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest 1996; 98: 345–351.
Acknowledgements
This study was funded by Grants DHSS/NIH/NIA 5R21 AG21560, NIH Grant R01DK073665 and Norman Coplon Research Grant (DSCR), and DHSS/NIH/NCRR/GCRC 5MO1 RR00997.
The authors express sincere gratitude to the study participants, Linda Friesen, Rosemary Wold, Elizabeth, and GCRC inpatient and outpatient nurses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Shah, V., Scariano, J., Waters, D. et al. Mitochondrial DNA deletion and sarcopenia. Genet Med 11, 147–152 (2009). https://doi.org/10.1097/GIM.0b013e31819307a2
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31819307a2
