
Abstract
Abstract: MEN 1 is a rare hereditary cancer syndrome which manifests a variety of endocrine and non-endocrine neoplasms and lesions. Growing knowledge of this condition in both its molecular genetic underpinnings and its clinical implications have affected the entire spectrum of the clinical management of MEN patients. The MEN1 gene is a tumor suppressor gene, and mutations in it account for the development of the MEN1 clinical syndrome through impairment of several cell functions, such as cell proliferations, cell growth control, apoptosis, DNA replication and repair, gene expression, transcriptional machinery control, and hormone secretion. Currently, DNA testing makes possible the early identification of germline mutations in asymptomatic mutation carriers. The ever increrasing combination of genetic and clinical tools will allow early detection of MEN1-associated neoplasms, potentially improving clinical outcomes and quality of life for both affected patients and their relatives.
Similar content being viewed by others

Main
The multiple endocrine neoplasia type 1 (MEN1) syndrome (MIM#131100) was originally described at the beginning of the past century at the autopsy of a patient concurrently exhibiting acromegaly and multiglandular parathyroid abnormalities.1 At the end of the 1920s, the presence of the “classical” P-triad (Parathyroid, Pancreatic, and Pituitary adenomas) was reported in a patient.2 In 1953, a first review of related syndromes in 14 cases was published.3 Up to this period, the existence of the syndrome as a distinct clinical entity had not been assumed or suspected. In 1954, Wermer4 was the first to suggest that the P-triad could represent a trait inherited in an autosomal dominant manner with high penetrance. This insight is honored by also referring to MEN1 as Wermer syndrome.
However, it was not until the 1960s and later, when radioimmunoassays for hormonal and peptidic products of the affected endocrine tissues were developed,5–7 that more careful definitions of the details of the clinical features of the disorder could be made. The clinical management of individuals with MEN1 has greatly benefited from the advances in knowledge of either surgical or pharmacological interventions or molecular mechanisms underlying the tumorigenesis of the sporadic, nonsyndromic, and counterparts of those endocrine tumors known to be associated with the MEN1 syndrome.8–10
Today, it has been well established that MEN1 syndrome may present with a varying combination of more than 20 endocrine and nonendocrine tumors, other than combinations of parathyroid adenoma(s), gastrinoma(s), and prolactinoma.11
In fact, other endocrine and nonendocrine neoplasms such as foregut carcinoid, central nervous system (CNS) tumors, leyomiomas, lipomas, and particularly skin tumors have also been demonstrated to be common. Furthermore, other endocrine and nonendocrine tissues can be affected but with a lower frequency (Table 1). In this review, we update genetic and clinical aspects.
FAMILIAL/SIMPLEX MEN1 CASES AND THEIR DEFINITION
Despite extreme variation in the range of tumors which occur, MEN1 can be practically regarded as a disease with two of three principal MEN1-related endocrine tumors (parathyroid, pituitary and gastro-entero-pancreatic [GEP] tract [neuro]endocrine tumors).
The familial form of MEN1 syndrome occurs with a significantly higher frequency, approximately 90%, than the simplex form (only one affected individual without evidence of a positive familial history), that occurs approximately in 10% of cases. The familial form is defined as MEN1 syndrome in an individual who has either at least one first-degree relative with at least one of the main endocrine tumors or only one organ involvement and a MEN1 disease-causing germline mutation.12
CLINICAL ASPECTS OF MEN1 SYNDROME
Clinical diagnosis
Diagnosis of MEN1-associated endocrine tumors is easier if there is evidence of hormonal overproduction by the tumor than through identification of symptoms related to the growth of the tumor itself.
MEN1-associated endocrine tumors
Most of the tumors in MEN1 are benign, even if some GEP neuroendocrine tumors (mainly gastrinomas) and foregut carcinoids are often malignant. Although often regarded as a treatable endocrinopathy, MEN1 can also be regarded as a cancer syndrome lacking effective prevention or cure for associated malignancies. Tables 1 and 2 describe the MEN1-associated endocrine and nonendocrine tumors and their penetrance estimated at 40 years of age, Table 3 reports the salient clinical manifestations of MEN1-associated (neuro)endocrine tumors.
Parathyroid tumors
Primary hyperparathyroidism (PHPT) is the main MEN1-associated endocrinopathy occurring in 90% of individuals between ages 20 and 25 years and manifests as hypercalcemia in 100% of affected individuals by age 50 years (high penetrance); hypercalcemia may cause lethargy, depression, confusion, anorexia, constipation, nausea, vomiting, diuresis, dehydration, hypercalciuria, kidney stones, increased bone resorption and increased fracture risk, hypertension, and shortened QT interval. However, MEN1-PHPT is frequently asymptomatic for a long period of time.13
The MEN1 parathyroid disease is a multiglandular disorder with enlargement of all the parathyroid glands occurring asynchronically and asymmetrically.14
Risk of malignant progression
Malignant progression of parathyroid tumors is not a frequent feature of MEN1 syndrome, even if some reports described the presence of parathyroid carcinoma in patients with MEN1.15–17
Pituitary tumors
Pituitary tumors may occur as the first clinical manifestation of MEN1 syndrome in 25% of simplex and in 10% of familial cases. Verges et al.18 reported that pituitary involvement was the initial manifestation of MEN1 syndrome in 17% of individuals and that pituitary adenomas were significantly more frequent in women than in men. The occurrence of anterior pituitary tumors in MEN1 syndrome may range between 10 and 60% depending on the study.12 Between 6512 and 85%18 of pituitary tumors in MEN1 syndrome are represented by macroadenomas and plurihormonal adenomas can occur more frequently than monohormonal adenomas in MEN1. A patient with MEN1 and two simultaneously occurring pituitary tumors (one gonadotrope macroadenoma and one corticotrope microadenoma) has been also reported.19 Recently, an unusual association between the growth hormone (GH) and prolactin (PRL) plurihormonality with follicle-stimulating hormone, luteinizing hormone, or adrenocorticotropic hormone (ACTH) has been reported to be more frequent in MEN1 tumors than non-MEN1 controls.20
Pituitary tumors are mainly functioning tumors and prolactinoma is the most common among them, manifesting as oligomenorrhea/amenorrhea and galactorrhea in women and sexual dysfunction and, more rarely, gynecomastia in men. In some families, including four families from Newfoundland reported as the so-called MEN1Burin variant, prolactinoma is unusually common.21 Hypercortisolism occurs in ACTH-secreting tumors22,23 and gigantism/acromegaly occur in children/adults with GH-secreting tumors.24 Symptoms such as nerve compression, headache, and hypopituitarism may also result from pituitary mass effects.25,26
Risk of malignant progression
Although the MEN1 French-Belgian Registry reported that 32% of pituitary macroadenomas were locally invasive, malignant degeneration of MEN1-associated pituitary tumors is an infrequent event.18 However, recently, it has been described a metastatic PRL-oma, presenting as a cervical spinal cord tumor, in one MEN1 affected individual.27
Well-differentiated endocrine tumors of the GEP tract
They include neuroendocrine tumors of the stomach, duodenum, pancreas, and the intestinal tract.28 These tumors may clinically manifest as Zollinger-Ellison syndrome (ZES) resulting from gastrinoma(s); hypoglycemia resulting from an insulinoma; hyperglycemia, anorexia, glossitis, anemia, diarrhea, venous thrombosis, and skin rash resulting from a glucagonoma; and watery diarrhea, hypokalemia, and achlorhydria syndrome resulting from a vasoactive intestinal peptide (VIP) secreting tumor, VIPoma.
Adrenocortical tumors are frequently nonfunctioning neoplasms but sometimes are associated with primary hypercortisolism or hyperaldosteronism.
Carcinoid tumors are generally nonhormone-secreting tumors, and they manifest as a large mass after 50 years of age.
Gastrinoma(s)
Approximately 40% of individuals with MEN1 syndrome have gastrinoma(s), which manifests as ZES. Clinical findings can include upper abdominal pain, diarrhea, esophageal reflux, and acid-peptic ulcers; if not properly diagnosed or treated, as occurred in the past, ulcer perforation can develop from hypergastrinemia, even without prior symptoms. Heartburn and weight loss, which are less commonly reported, may be also described. ZES-associated hypergastrinemia may result in multiple duodenal ulcers; epigastric pain generally occurs 2 or more hours after meals or at night and may be relieved by eating. However, the pain may also be in the right upper quadrant, chest, or back. Vomiting may be related to partial or complete gastric outlet obstruction; hematemesis or melena may result from gastrointestinal bleeding. ZES usually occurs before 40 years of age.29 In the MEN1Burin variant, the prevalence of gastrinoma is lower than in classical MEN1.30
At pathology, multiple small (<1 cm in diameter) gastrinomas are typically observed in the duodenal submucosa. In particular, more than 80% of MEN1 gastrinomas are commonly found within the first and second duodenum portion,31 likely arising from multifocal/hyperplastic proliferations of gastrin-producing cells. MEN1 duodenal gastrinomas are associated with diffuse hyperplastic changes of gastrin cells and multicenter microtumors (<1 mm) producing gastrina.32
About 50% of duodenal microgastrinomas exhibit loss of heterozygosity at MEN1 locus and thus might represent the initial tumors.33 In fact, multifocal duodenal endocrine tumors are presumably arising by independent clonal events after the germline MEN1 mutation.34 Such precursor lesions are not reported in sporadic, non-MEN1, gastrinomas.33 Pancreatic gastrinomas are extremely rare in patients with MEN1.35
Risk of malignant progression
MEN1-gastrinomas usually include a malignant component and in approximately 50% of subjects with MEN1 has metastasized before diagnosis.12,31,36 Individuals with liver metastases have a poor prognosis for survival; this contrasts with nodal metastases, which do not seem to negatively influence prognosis.
Insulinoma
The age of onset of MEN1-insulinoma, clinically evidenced as recurrent fasting hypoglycaemic crises, is generally one decade earlier than the sporadic counterpart.37
At pathology, generally a single tumor occurs in the setting of multiple islet macroadenomas.12 Tumors responsible for hyperinsulinism are usually about 1–4 cm in diameter.38
Risk of malignant progression
MEN1-insulinomas are almost always benign.39
Rare functioning GEP tumors
They include VIPomas, glucagonomas, somatostatinomas, or GH-releasing factoromas, usually exhibiting a large diameter (>3 cm).35 Recently, cervical metastases of glucagonoma in a patient with MEN1 have been reported. After a pancreatoduodenectomy, the patient recovered well and remained asymptomatic.40
Nonsecreting GEP tract tumors
They are frequent in MEN1 syndrome. A prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreatic tumors in patients with MEN1 suggested that their frequency is higher (55%) than previously thought.41 The penetrance of these tumors has been reported to be 34% at the age of 50 years in patients with MEN1 from the French Endocrine Tumor Study Group, indicating them as the most frequent pancreaticoduodenal tumor in patients with MEN1. Average life expectancy for patients with these tumors was shorter than that for patients with MEN1 who did not have pancreaticoduodenal tumors.42
Risk of malignant progression
Interestingly, a long-term follow-up study in Japanese patients with MEN1 revealed that nonfunctioning pancreatic tumors, smaller than 20 mm in diameter, did not show any apparent growth over a long monitoring period without metastasizing to regional lymph nodes or the liver.43
Carcinoid tumors
Thymic, bronchial, and type II gastric enterochromaffin-like carcinoids occur approximately in 10% of individuals with MEN1 syndrome. They rarely oversecrete ACTH, calcitonin, or GH-releasing factor; similarly, they rarely oversecrete serotonin or histamine and rarely cause the carcinoid syndrome. However, GH production by a thymic carcinoid causing acromegaly has been reported44 as also Cushing syndrome due to ACTH-producing thymic carcinoids.45,46 The clinical course of carcinoid tumors is often indolent, but it can also be aggressive and resistant to therapy.47
Thymic carcinoid tumors are generally a late manifestation of MEN1 syndrome because no affected individuals had thymic carcinoid as the initial manifestation of MEN1 syndrome.48
Risk of malignant progression
They commonly present at an advanced stage as a large invasive mass and are more prevalent in men than in women.49 The MEN1-associated thymic carcinoids tend to be aggressive and highly lethal, particularly in men who are smokers.48,50,51 Recently, a spinal metastasis of a carcinoid tumor has been also reported in a patient with MEN1.52
On the other hand, bronchial carcinoids are more prevalent in women than in men. Bronchial carcinoids, often multicentric, may exhibit both synchronous and metasynchronous occurrence. In contrast to thymic carcinoids, most bronchial carcinoids usually behave indolently, albeit with the potential for local mass effect, metastasis, and recurrence after their resection.53
Gastric carcinoids have a mean age at diagnosis of 50 years. In up to 15% of individuals with MEN1 syndrome, they are recognized incidentally during endoscopy.54–56
Adrenocortical tumors
In MEN1, they are generally nonfunctioning but rarely may be associated with elevated serum concentrations of cortisol or aldosterone.57–59 Mono- or bilateral adrenocortical tumors are present in 20–40% of individuals with MEN1 syndrome.60
Risk of malignant progression
Their cancer progression risk is very low. Recently, a prospective study confirmed that adrenal lesions were invariably nonfunctional, and no malignant transformation was reported.61
MEN1-associated nonendocrine tumors
Nonendocrine tumors associated with MEN1 syndrome include skin tumors such as facial angiofibromas, collagenomas, lipomas, CNS tumors such as meningiomas, ependymomas, and leiomyomas.13 Table 2 describes the MEN1-associated endocrine tumors and their penetrance estimated at the age of 40 years.
Skin tumors
Facial angiofibromas are benign tumors, comprised of blood vessels and connective tissue, consisting of acneiform papules that do not regress and may extend across the vermillion border of the lips.
Collagenomas are generally multiple, skin-colored, sometimes hypopigmented, cutaneous nodules, symmetrically arranged on the trunk, neck, and upper limbs. They are typically asymptomatic, rounded, and firm-elastic, from a few millimeters to several centimeters in size.
Lipomas consist of multiple benign fatty tissue tumors found anywhere that fat is located. They are generally subcutaneous or, rarely, visceral.
Because multiple facial angiofibromas have been reported in 88%, collagenomas in 72%, café au lait macules in 38%, lipomas in 34%, confetti-like hypopigmented macules in 6%, and multiple gingival papules in 6%, it has been suggested that these cutaneous findings may be helpful in the diagnosis of individuals with MEN1 syndrome before manifestations of hormone-secreting tumors appear.62,63 Although a rapid growth of protuberant collagenomas seems to be unusual in MEN1, a patient with MEN1 exhibiting rapid growth of multiple collagenomas, after pancreatic enucleation of a VIPoma and excision of multiple pancreatic masses, has been reported, suggesting that MEN1 be considered in patients with apparent eruptive collagenoma(s).64
CNS tumors
The presence of a meningioma has been reported in 8% of 74 individuals with MEN163; the meningiomas were mainly asymptomatic and 60% showed no growth. Ependymoma has been described in 1% of subjects with MEN1.65
Leiomyomas
They are benign neoplasms derived from smooth (nonstriated) muscle.66,67
GENETIC ASPECTS OF MEN1 SYNDROME
Chromosomal location of the MEN1 gene and model for the related tumorigenesis
Combined studies of both linkage in MEN1 kindreds,68 and microdeletion analysis in tumors68–70 demonstrated that the MEN1 trait was closely linked to PYGM gene, encoding for muscle phosphorylase, at chromosome 11q13. The proposed model for tumorigenesis in familial MEN1 is, according to Knudson's “two hits” hypothesis,71 a mutated MEN1 copy is inherited at the germline level from the affected parent (first hit), whereas the wild-type copy, inherited from the healthy parent, is lost at the somatic level (second hit), resulting in a tumor. This mechanism is supported by the observation of loss of heterozygosity or allelic loss when tumor DNA is compared with constitutive DNA (Fig. 1). These findings strongly suggested gene inactivation as the mechanism of tumorigenesis for MEN1.

Example of a loss of heterozygosity or allelic loss at 11q13 region in DNA from a MEN1-associated tumor (T) with respect to the retention of both alleles in the constitutive (C) DNA. The lost allele derives from the healthy, unaffected, parent. Consequently, such a finding suggests the loss of a tumor suppressor gene.
MEN1 gene and its mutations
In 1997, the MEN1 gene was identified by positional cloning72,73 and is the only gene known to be associated with MEN1 syndrome.
The gene spans 9 kb and consists of 10 exons with an 1830-bp coding region (Fig. 2) that encodes a novel 610-amino acid protein, referred to as menin. The first exon and part of exon 10 are untranslated. A major transcript of 2.8 kb has been described in a large variety of human tissues (pancreas, thymus, adrenal glands, thyroid, testis, leukocytes, heart, brain, lung, muscle, small intestine, liver, and kidney).73 Moreover, an additional transcript of approximately 4 kb has been detected in pancreas and thymus, suggesting a tissue-specific alternative splicing.73

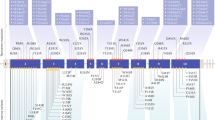
Schematic representation of the MEN1 gene. White boxes indicate exons, horizontal bars indicate introns, gray boxes represent untranslated regions (UTRs). Vertical bold black bars (A-I) above the gene represent the site of the nine most common germline mutations, potential mutational hot spots (frequency > 1.5%), length of bars indicates the mutation frequency according to the scale shown on the left (see also Ref. 74). Vertical black bar below the gene indicate the founder mutations in very large MEN1 families (see also Ref. 111).
More than 1000 different germline MEN1 mutations are scattered in and around the open reading frame without significant clustering that corresponds to functional domains of the protein.18,72,74–93 Of all mutations, about 25% are nonsense, 45% are deletions, 15% are insertions, <5% are splice site mutations, and 10% are missense mutations. Twenty-four normal variants (polymorphisms) have been described.74 Most germline or somatic mutations in the MEN1 gene predict truncation or absence of encoded menin. Neither the finding of a tumor suppressor mechanism nor the identification of binding partners has established the ultimate pathways of menin action in normal tissues or in tumors.93
MEN1 germline mutations are identified by sequencing analysis in about 80–90% of probands with familial MEN1 syndrome12 and about 65% of individuals with simplex MEN1 syndrome (88 < 32). Approximately 45% of germline mutations detected by a sequence analysis are small deletions and approximately 15% are small insertions.12 Intronic MEN1 mutations have been also reported in the literature, representing splicing-affecting genomic variants that are likely to be of significance in the 10% of patients with MEN1 who do not have coding region mutations.94,95 Recently, a new intronic mutation, at intron 3 of the MEN1 gene, has been described to be associated with PRL-oma, decreased familial penetrance, and variable effects on MEN1 mRNA and menin.96
Different mutation detection rates have been reported in different series, and the likelihood of detecting a MEN1 mutation is higher in individuals with more main tumors (parathyroid, pancreatic, and pituitary), especially those from families with hyperparathyroidism and pancreatic islet tumors.97,98 In particular, it has been described that pancreatic manifestations were significantly linked with the probability of mutation and within a sporadic context with at least two established manifestations of MEN1, the overall probability of identifying a mutation was 26%, warranting MEN1 genotypic analysis.99 MEN1 genetic screening should also be offered to patients with PHPT or gastrinomas after thorough investigation into the family history, whereas sporadic carcinoid tumors or primary prolactinomas are rarely associated with germline MEN1 mutations.100
Simplex MEN1 cases are less likely to test positive than familial cases, in part because some of these simplex cases are caused by somatic mosaicism.98 Individuals who have a single MEN1-related tumor and no family history of MEN1 syndrome rarely have germline MEN1 mutations.97
Approximately, 1–3% of MEN1 germline mutations may consist of large deletions, detectable by a Southern blot analysis or other gene dosage procedures (i.e., polymerase chain reaction based).97,98,101–103 Multiplex ligation-dependent probe amplification assay may detect large deletions (4%) as germline mutations in MEN1.104 Through polymorphism analyses, gene dose assay and nucleotide sequencing, a large germline deletion of approximately 29 kbp spanning the whole MEN1 gene has been identified in one patient with a positive familial history for MEN1 whose germline MEN1 mutation was undetectable by a conventional sequencing analysis.105
Moreover, through the application of a forensic technique for ancient DNA by 11q13 haplotype-based analysis, in eight living and one deceased member of a MEN1 Italian family lacking of detection of any germline mutation of the MEN1 gene, it has been possible to identify the familial disease-associated haplotype, demonstrating that MEN1 disease haplotype family history can be reconstructed even when one or more family members are deceased.106
Finally, linkage analysis may be used in certain families if a disease-causing mutation is not identified using a sequence analysis or targeted mutation analysis or multiplex ligation-dependent probe amplification analysis.
Founding MEN1 mutations
To date, 13 MEN1 founding mutations have been described in European (France, Finland, and Sweden), North American (United States and Canada), South American (Brazil), and Oceania (Australia) kindreds107–114 (Fig. 2). Founder effect has been reported as the result of combination of either genetic or clinical studies. Genealogical data, common geographic origin, common MEN1 mutations, and common haplotypes were described for each MEN1 family.
MEN1 variants and phenocopies
MEN1 variants manifest as different clinical consequences from mutations in the MEN1 gene. For example, familial isolated hyperparathyroidism, in which germline mutations in MEN1 gene have been reported, only results in parathyroid dysregulation. On the other hand, MEN1 phenocopies occur when a clinical presentation resembles MEN1 but does not result from an alteration in the MEN1 gene. An example of such a situation would be a family with familial isolated hyperparathyroidism who has negative MEN1 mutation analysis but a mutation in a different familial hyperparathyroidism related gene, such as the calcium sensing receptor gene (CaSR).115
Recently, germline mutations in the CDKN1B/p27 gene, encoding a protein belonging to the Cip/Kip family members of cyclin-dependent kinase inhibitor (CDKI) family, have been reported116 in an MEN1-like state featured by familial occurrence of PHPT and pituitary tumors, known as MEN4 syndrome. Such heterogeneity highlights the need for caution in interpretation of negative mutational analyses. Agarwal et al.117 have demonstrated that germline CDKN1B/p27 mutations are associated in a small, but important, fraction of cases with MEN1 and related states.
Menin is mainly located in the nucleus93,118 and its C-terminal part contains sequences, overlapping with nuclear localization domains, essential for the regulation of gene expression.118 It has been recently demonstrated that a splicing mutation of MEN1 alters the splice acceptor site of intron 9 that promotes an incorrect splicing, generating aberrant proteins without the nuclear localization signals necessary for the normal menin translocation to the nucleus.119 Menin is widely expressed and may play different roles in different tissues. It is probably involved in the physiological regulation of several cell functions, including DNA replication and repair, as also in transcriptional machinery.
Menin may inhibit JunD-mediated transcriptional activation, because studies of deletion mutants have shown the existence of interacting regions of both proteins. Menin could inhibit JunD-mediated transcription by modification of chromatin structure recruiting a specific histone deacetylase targeted to a promoter by binding JunD. Moreover, when compared with controls, lymphocytes from individuals with a heterozygous MEN1 mutation show both premature division of the centromere and hypersensitivity to alkylating agents. Thus, menin could be a negative regulator of cell proliferation after DNA damage. Menin has been reported to interact with a multitude of proteins, other than JunD, including SMAD family members, Pem, NFκB, FANCD2, RPA2, NMMHC II-A, GFAP, vimentin, p53, and Hsp70, although to date none of the interacting partners have been directly proved important in MEN1 pathophysiology.120–137 It has been hypothesized that menin may mediate its tumor suppressor action by regulating histone methylation in promoters of HOX genes and/or CDKN2C/p18, CDKN1B/p27, and possibly other CDKIs.138,139 Consistent with this hypothesis, H3 K4 methylation and expression of p18 and p27 were shown to be dependent on menin in pancreatic islets.139 An additional clue for a role of p18 and p27 in MEN1 pathophysiology comes from the studies in knockout mice,140 in which the simultaneous loss of p18 and p27, CDKI, leads to a tumor spectrum similar to that in human MEN1 and MEN2 patients, including tumors in the pituitary, parathyroid, thyroid, endocrine pancreas, stomach, and duodenum. Moreover, a physiological role for menin has been postulated either in bone development or hematopoiesis, also regulating lymphoid progenitors.124,129,136,141 However, it still remains to be delineated what determines the tissue-specific activities of menin and whether the menin–MLL–Hox pathway has a role in suppressing tumorigenesis in endocrine organ.
Through serial analysis of chromatin occupancy, a method combining chromatin immunoprecipitation with serial analysis of gene expression, hundreds of menin-occupied genomic sites were identified in promoter regions, near the 3′ end of genes, or inside genes, extending other data about menin recruitments to many sites of transcriptional activity. Moreover, a large number of menin-occupied sites were located outside known gene regions.131 Interestingly, it has been shown that wild-type menin, but not MEN1 disease-derived mutants, physically interacts with p53 and that ectopic menin expression in insulinoma cells enhances γ-irradiation-induced apoptosis, p21 expression and proliferation inhibition. Because activated p53 normally stimulates transcription of p21, a CDKI and an inhibitor of cell proliferation, and also multiple Bcl-2 homology domain-containing proapoptotic proteins such as PUMA, these findings could explain how menin, at least in part, regulates proliferation and apoptosis of endocrine cell through an interaction with p53.142 However, although many menin-interacting pathways have been described, it is highly likely that only a few basilar molecular pathways are involved in menin-dependent tumorigenesis.
Genotype-phenotype correlations
With the exception of the MEN1Burin variant, a phenotype reported in four kindreds from Newfoundland and in one from Mauritius, where a higher prevalence for prolactinoma has been described,21 no genotype-phenotype correlations have been clearly reported in MEN1 syndrome,74,90,91,110 even if a statistically not significant trend for a high prevalence of truncating mutations in MEN1-related thymic carcinoids has been described.143
Genetic counseling and DNA test
As mentioned earlier, MEN1 syndrome is inherited in an autosomal dominant manner. Consequently, each child of an individual with MEN1 syndrome has a 50% chance of inheriting the disease-causing mutation. In general, the risk to other family members depends on the status of the proband's parents. If a parent is found to be affected and/or to have a disease-causing mutation, his or her family members are at risk.
Although approximately 90% of individuals diagnosed with MEN1 syndrome have an affected parent, the family history may seem to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent.13 The penetrance of this syndrome approaches 100% with increasing age and with a variable expression (different clinical phenotypes in affected members within a MEN1 kindred, in terms of tumor localization, onset age, and clinical aggressiveness). Variable expression is frequently observed among members from different MEN1 families sharing a common MEN1 mutation. Because there are no definitive data on cell tissue-specific interactions between menin and proteins involved in regulating the growth and cellular functions, or on the existence of gene modifiers of the age of onset, such a variable expression cannot be currently explained. However, according to the above described roles played by menin in regulation of transcription, genome stability, cell division and proliferation, in the control of DNA repair and genome stability, it could be hypothesized that accumulation of randomly occurring genetic damage at the cellular level could foster tumorigenesis in different organs or tissue.
On the other hand, the possible existence of cell-specific factors for menin could explain a permissive endocrine environment for MEN1 tumorigenesis. As recently reported in an animal model, ability of menin in the maintenance of both cellular and microenvironment integrity could account for the endocrine-restrictive nature of the MEN1 syndrome.144
Counseling of patients and closely related family members has a central role in the management of hereditary diseases, such as MEN1 syndrome. In fact, subjects with a genetic predisposition for this tumor syndrome may greatly benefit from early identification, especially at a presymptomatic stage by a DNA analysis. After the genetic status is disclosed, MEN1 gene carriers have to be followed by a periodic clinical tumor surveillance for expression of the disease as well as recurrence after treatment or progression of the disease, because the knowledge about carrier status enables early diagnosis and intervention, as reported in the literature.13,145 Moreover, no less important is the fact that knowledge decreases uncertainty and anxiety and assists in making decisions about lifetime plans.
It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk. The latter comprises subjects who have inherited the MEN1 mutation from one affected parent or are the relatives of subjects clinically defined as suffering from MEN1 and in which the germline mutation has not been identified.
The advantages of DNA analysis are that it requires a single blood sample and does not need to be repeated in theory, because the analysis is independent of the age of the individual and provides an objective result. Requests for prenatal testing for conditions such as MEN1 syndrome that do not affect intellect and have some treatment available are not common, and an universal consensus on the opportunity to perform such a diagnosis in MEN1 does not exist. Moreover, there may be no role for genetic testing if there is no planned pregnancy in the future.
DNA test in MEN1 may be undertaken in children within the first decade because tumors such as insulinoma and pituitary adenomas have developed in some children by the age of 5 years.24,145 Moreover, it has been reported that genetically diagnosed patients already harbored manifestations at the time of diagnosis confirming that screening for a MEN1 mutation should be done at an early age.146
Unfortunately, the great diversity together with widely scattered locations of the MEN1 mutations74 and a lack of genotype–phenotype correlation, make such mutational screening time consuming, arduous, and expensive.74 An appropriate intervention in the form of clinical-biochemical testing, or treatment, or both, has been considered.145 Tables 4 and 5 summarize a minimal clinical surveillance for subjects known to have MEN1 on genetic and/or clinical basis (Table 4) and for individuals at 50% risk of having MEN1 syndrome whose genetic status is unknown (Table 5). In many centers following subjects with MEN1, there is an integrated program of both mutational analysis, to identify mutant gene carriers, and clinical-biochemical screening, to detect the development of tumors.145 Although currently a DNA test identifying an individual as a mutant gene carrier does not usually lead to immediate medical or surgical treatment, it suggests a precocious and frequent clinical screening, whereas a DNA result indicating that an individual is not at risk will suggest no further clinical investigations. However, because of the current inability to individually predict tumor penetrance and malignancy, lifelong follow-up of MEN1 carriers is strongly recommended to prevent tumor morbidity.
Finally, we believe that it is important that the medical counselor has to be involved either in clinical network or in informing the family doctor. Moreover, it is important that the medical counselor be familiar with both basic and clinical research, especially in light of new potential therapeutic strategies that may be offered soon in patients with MEN1.
MORBIDITY AND MORTALITY OF MEN1 SYNDROME
In the past 10 years, the improved knowledge of MEN1 syndrome-associated clinical manifestations, facilitated by an early diagnosis of MEN1 syndrome-associated tumors, and treatment of metabolic complications of MEN1, has virtually eliminated ZES and/or complicated PHPT as causes of death. Interestingly, according to a study performed in a single large MEN1 pedigree from Oceania, diabetes and impaired fasting glucose significantly occurred more frequently among patients with MEN1 than controls and was associated with uncontrolled hyperparathyroidism and evidence of enteropancreatic hyperstimulation.147 Although such findings need to be repeated in subjects with MEN1 from other ethnicities, they may suggest physicians to look more carefully for glucose homeostasis impairment in all patients with MEN1.
Nonetheless, individuals with MEN1 syndrome have a significantly increased risk of premature death, justifying surveillance of those with MEN1 mutations and/or a family history of MEN1 syndrome.148 Longer life expectancy in MEN1 syndrome is likely to result in a rising cumulative morbidity and mortality from MEN1 syndrome-associated malignancies, which currently account for approximately 30% of deaths in MEN1 syndrome.
Gene testing decreased the morbidity and mortality associated with MEN1. In a multicenter study on 258 MEN1 gene carriers, it has been reported “as a result of differential tumor detection, MEN1 carriers born during the second half of the 20th century tend to have their tumors diagnosed earlier than carriers of the same age born in the first half.”149
A prospective clinical study on MEN1 mutant gene carriers revealed that biochemical evidence of neoplasia could be identified on average 10 years after evidence of clinical disease, allowing for early surgery. Thus, mutation positive individuals should undergo a focused surveillance for early identification of potentially malignant neuroendocrine tumors because of the potential for morbidity and mortality related to the syndrome.150
CONCLUSIONS
Since the discovery of the MEN1 gene, changes in clinical management have occurred. Unfortunately, neither the finding of a tumor suppressor mechanism nor the identification of binding partners has established the ultimate pathways of menin action in normal tissues or in tumors.93 However, identification of several menin interacting partners strongly contribute to increase the knowledge of the pathophysiological actions of menin, but more studies are necessary to clearly elucidate both the molecular mechanisms underlying MEN1-associated tumorigenesis and the role(s) played by menin in tumor development. In the near future, there are prospects for novel treatments based on DNA, RNA, or even other small molecules. Further studies on the intricate molecular pathway networks of menin will be helpful to design novel therapeutic modalities.
REFERENCES
Erdheim J . Zur normalen und pathologischen histologie der glandula thyroidea, parathyroidea, und hypophysis. Beitr Pathol Anat 1903; 33: 158–236.
Cushing H, Davidoff LM . Studies in acromegaly V. The pathological finding in four autopsied cases of acromegaly with a discussion of their significance. Monograph, New York, NY: Rockefeller Institute for Medical Research, 1927; 22.
Underdahl LO, Woolner LB, Black BM . Multiple endocrine adenomas: report of eight cases in which the parathyroids, pituitary and pancreatic islets were involved. J Clin Endocrinol Metab 1953; 13: 20–25.
Wermer P . Genetic aspects of adenomatosis of endocrine glands. Am J Med 1954; 16: 363.
Yalow RS . Radioimmunoassay: a probe for the fine structure of biologic systems. Science 1978; 200: 1236–1245.
Malarkey WB . Prolactin and the diagnosis of pituitary tumors. Annu Rev Med 1979; 30: 249–258.
Nussbaum SR, Potts JT Jr . Immunoassays for parathyroid hormone 1–84 in the diagnosis of hyperparathyroidism. J Bone Miner Res 1991; 6: S43–S50.
Albright F, Ellsworth R, Loriaux DL, eds. Uncharted Seas. Portland, OR: Kalmia Press, 1990.
Bevan JS, Webster J, Burke W, Scanlon MF . Dopamine agonists and pituitary tumor shrinkage. Endocr Rev 1992; 13: 220–240.
Schmid JR, Labhart A, Rossier PH . Relationship of multiple endocrine adenomas to the syndrome of ulcerogenic islet cell adenomas (Zollinger-Ellison). Occurrence of both syndromes in one family. Am J Med 1961; 31: 343–348.
Gagel RF, Marx SJ . Multiple endocrine neoplasia. In: Larsen PR, Kronenberg H, Melmed S, Polonsky K, editors. Williams textbook of endocrinology, 11th ed. Orlando, FL: WB Saunders & Company, 2007.
Brandi ML, Bordi C, Tonelli F, et al. Multiple endocrine neoplasia type 1. In: Bilezikian JP, Raisz GA, Rodan LG, editors. Principles of bone biology, 3rd ed. San Diego, CA: Academic Press, 2008: 1345–1374. Chapter 62.
Falchetti A, Marini F, Brandi ML . Multiple endocrine neoplasia type 1. In: GeneReviews at GeneTests: Medical Genetics Information Resource [database online]. Copyright, University of Washington, Seattle, 1997–2005. Available at: http://www.genetests.org. Accessed August 2005.
Marx SJ . Multiple endocrine neoplasia type 1. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease, 8th ed. New York: McGraw-Hill, 2001.
Sato M, Miyauchi A, Namihira H, et al. A newly recognized germline mutation of MEN1 gene identified in a patient with parathyroid adenoma and carcinoma. Endocrine 2000; 12: 223–226.
Dionisi S, Minisola S, Pepe J, et al. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc 2002; 77: 866–869.
Agha A, Carpenter R, Bhattacharya S, Edmonson SJ, Carlsen E, Monson JP . Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: two case reports of an unrecognised entity. J Endocrinol Invest 2007; 30: 145–149.
Verges B, Boureille F, Goudet P, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002; 87: 457–465.
Al Brahim NY, Rambaldini G, Ezzat S, Asa SL . Complex endocrinopathies in MEN-1: diagnostic dilemmas in endocrine oncology. Endocr Pathol 2007; 18: 37–41.
Trouillas J, Labat-Moleur F, Sturm N, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol 2008; 32: 534–543.
Farid NR, Buehler S, Russell NA, Maroun FB, Allerdice P, Smyth HS . Prolactinomas in familial multiple endocrine neoplasia syndrome type I. Relationship to HLA and carcinoid tumors. Am J Med 1980; 69: 874–880.
Rix M, Hertel NT, Nielsen FC, et al. Cushing's disease in childhood as the first manifestation of multiple endocrine neoplasia syndrome type 1. Eur J Endocrinol 2004; 151: 709–715.
Matsuzaki LN, Canto-Costa MH, Hauache OM . Cushing's disease as the first clinical manifestation of multiple endocrine neoplasia type 1 (MEN1) associated with an R460X mutation of the MEN1 gene. Clin Endocrinol (Oxf) 2004; 60: 142–143.
Stratakis CA, Schussheim DH, Freedman SM, et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2000; 85: 4776–4780.
Yoshimoto K, Saito S . [Clinical characteristics in multiple endocrine neoplasia type 1 in Japan: a review of 106 patients]. Nippon Naibunpi Gakkai Zasshi 1991; 67: 764–774 ( in Japanese).
Carty SE, Helm AK, Amico JA, et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 1998; 124: 1106–1114.
Gordon MV, Varma D, McLean CA, Bittar RG, Burgess JR, Topliss DJ . Metastatic prolactinoma presenting as a cervical spinal cord tumour in multiple endocrine neoplasia type one (MEN-1). Clin Endocrinol (Oxf) 2007; 66: 150–152.
Kloppel G, Perren A, Heitz PU . The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann NY Acad Sci 2004; 1014: 13–1027.
Gibril F, Schumann M, Pace A, Jensen RT . Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 2004; 83: 43–83.
Hao W, Skarulis MC, Simonds WF, et al. Multiple endocrine neoplasia type 1 variant with frequent prolactinoma and rare gastrinoma. J Clin Endocrinol Metab 2004; 89: 3776–3784.
Hoffmann KM, Furukawa M, Jensen RT . Duodenal neuroendocrine tumors: classification, functional syndromes, diagnosis and medical treatment. Best Pract Res Clin Gastroenterol 2005; 19: 675–697.
Anlauf M, Perren A, Meyer CL, et al. Precursor lesions in patients with multiple endocrine neoplasia type 1-associated duodenal gastrinomas. Gastroenterology 2005; 128: 1187–1198.
Anlauf M, Perren A, Klöppel G . Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology 2007; 74: 279–284.
Anlauf M, Bauersfeld J, Raffel A, et al. Insulinomatosis: a multicentric insulinoma disease that frequently causes early recurrent hyperinsulinemic hypoglycemia. Am J Surg Pathol 2009; 33: 339–346.
Anlauf M, Perren A, Henopp T, et al. Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut 2007; 56: 637–644.
Fendrich V, Langer P, Waldmann J, Bartsch DK, Rothmund M . Management of sporadic and multiple endocrine neoplasia type 1 gastrinomas. Br J Surg 2007; 94: 1331–1341.
Marx SJ, Agarwal SK, Kester MB, et al. Multiple endocrine neoplasia type 1: clinical and genetic features of the hereditary endocrine neoplasias. Recent Prog Horm Res 1999; 54: 397–438.
Grama D, Skogseid B, Wilander E, et al. Pancreatic tumors in multiple endocrine neoplasia type 1: clinical presentation and surgical treatment. World J Surg 1992; 16: 611–618.
Mignon M, Ruszniewski P, Podevin P, et al. Current approach to the management of gastrinoma and insulinoma in adults with multiple endocrine neoplasia type I. World J Surg 1993; 17: 489–497.
Butte JM, Montero PH, Solar A, et al. Cervical metastases of glucagonoma in a patient with multiple endocrine neoplasia type 1: report of a case. Surg Today 2008; 38: 1137–1143.
Thomas-Marques L, Murat A, Delemer B, et al. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol 2006; 101: 266–273.
Triponez F, Dosseh D, Goudet P, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg 2006; 243: 265–272.
Sakurai A, Katai M, Yamashita K, Mori J, Fukushima Y, Hashizume K . Long-term follow-up of patients with multiple endocrine neoplasia type 1. Endocr J 2007; 54: 295–302.
Boix E, Pico A, Pinedo R, Aranda I, Kovacs K . Ectopic growth hormone-releasing hormone secretion by thymic carcinoid tumour. Clin Endocrinol (Oxf) 2002; 57: 131–134.
Takagi J, Otake K, Morishita M, et al. Multiple endocrine neoplasia type I and Cushing's syndrome due to an aggressive ACTH producing thymic carcinoid. Intern Med 2006; 45: 81–86.
Yano M, Fukai I, Kobayashi Y, et al. ACTH-secreting thymic carcinoid associated with multiple endocrine neoplasia type 1. Ann Thorac Surg 2006; 81: 366–368.
Schnirer II, Yao JC, Ajani JA . Carcinoid—a comprehensive review. Acta Oncol 2003; 42: 672–692.
Gibril F, Chen YJ, Schrump DS, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2003; 88: 1066–1081.
Teh BT, McArdle J, Chan SP, et al. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Medicine (Baltimore) 1997; 76: 21–29.
Ferolla P, Falchetti A, Filosso P, et al. Thymic Neuroendocrine Carcinoma (Carcinoid) in MEN1 Syndrome: the Italian series. J Clin Endocrinol Metab 2005; 90: 2603–2609.
Goudet P, Murat A, Cardot-Bauters C, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines). World J Surg 2009; 33: 1197–1207.
Tanabe M, Akatsuka K, Umeda S, et al. Metastasis of carcinoid to the arch of the axis in a multiple endocrine neoplasia patient: a case report. Spine J 2008; 8: 841–844.
Sachithanandan N, Harle RA, Burgess JR . Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer 2005; 103: 509–515.
Benya RV, Metz DC, Hijazi YJ, Fishbeyn VA, Pisegna JR, Jensen RT . Fine needle aspiration cytology of submucosal nodules in patients with Zollinger-Ellison syndrome. Am J Gastroenterol 1993; 88: 258–265.
Bordi C, D'Adda T, Azzoni C, Ferraro G . Pathogenesis of ECL cell tumors in humans. Yale J Biol Med 1998; 71: 273–284.
Gibril F, Reynolds JC, Lubensky IA, et al. Ability of somatostatin receptor scintigraphy to identify patients with gastric carcinoids: a prospective study. J Nucl Med 2000; 41: 1646–1656.
Beckers A, Abs R, Willems PJ, et al. Aldosterone-secreting adrenal adenoma as part of multiple endocrine neoplasia type 1 (MEN1): loss of heterozygosity for polymorphic chromosome 11 deoxyribonucleic acid markers, including the MEN1 locus. J Clin Endocrinol Metab 1992; 75: 564–570.
Skogseid B, Larsson C, Lindgren PG, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 1992; 75: 76–81.
Honda M, Tsukada T, Horiuchi T, et al. Primary hyperparathyroidism associated with aldosterone-producing adrenocortical adenoma and breast cancer: relation to MEN1 gene. Intern Med 2004; 43: 310–314.
Skogseid B, Rastad J, Gobl A, et al. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery 1995; 118: 1077–1082.
Waldmann J, Fendrich V, Habbe N, et al. Screening of patients with multiple endocrine neoplasia type 1 (MEN-1): a critical analysis of its value. World J Surg 2009; 33: 1208–1218.
Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M . Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 1997; 133: 853–857.
Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT . Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab 2004; 89: 5328–5336.
Xia Y, Darling TN . Rapidly growing collagenomas in multiple endocrine neoplasia type I. J Am Acad Dermatol 2007; 56: 877–880.
Kato H, Uchimura I, Morohoshi M, et al. Multiple endocrine neoplasia type 1 associated with spinal ependymoma. Intern Med 1996; 35: 285–289.
McKeeby JL, Li X, Zhuang Z, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol 2001; 159: 1121–1127.
Ikota H, Tanimoto A, Komatsu H, Ozawa Y, Matsushita H . Ureteral leiomyoma causing hydronephrosis in Type 1 multiple endocrine neoplasia. Pathol Int 2004; 54: 457–459.
Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M . Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988; 332: 85–87.
Friedman E, Sakaguchi K, Bale AE, et al. Clonality of parathyroid tumors in familial multiple endocrine neoplasia type 1. N Engl J Med 1989; 321: 213–218.
Emmert-Buck MR, Lubensky IA, Dong Q, et al. Localization of the multiple endocrine neoplasia type I (MEN1) gene based on tumor loss of heterozygosity analysis. Cancer Res 1997; 57: 1855–1858.
Knudson AG . Antioncogenes and human cancer. Proc Natl Acad Sci USA 1993; 90: 10914–10921.
Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997; 276: 404–407.
Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet 1997; 6: 1177–1183.
Lemos MC, Thakker RV . Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29: 22–32.
Agarwal SK, Kester MB, Debelenko LV, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 1997; 6: 1169–1175.
Heppner C, Kester MB, Agarwal SK, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet 1997; 16: 375–378.
Bassett JH, Forbes SA, Pannett AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998; 62: 232–244.
Carling T, Correa P, Hessman O, et al. Parathyroid MEN1 gene mutations in relation to clinical characteristics of nonfamilial primary hyperparathyroidism. J Clin Endocrinol Metab 1998; 83: 2960–2963.
Farnebo F, Teh BT, Kytölä S, et al. Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab 1998; 83: 2627–2630.
Giraud S, Zhang CX, Serova-Sinilnikova O, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 1998; 63: 455–467.
Sato M, Matsubara S, Miyauchi A, et al. Identification of five novel germline mutations of the MEN1 gene in Japanese multiple endocrine neoplasia type 1 (MEN1) families. J Med Genet 1998; 35: 915–919.
Teh BT, Kytölä S, Farnebo F, et al. Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab 1998; 83: 2621–2626.
Vortmeyer AO, Böni R, Pak E, Pack S, Zhuang Z . Multiple endocrine neoplasia 1 gene alterations in MEN1-associated and sporadic lipomas. J Natl Cancer Inst 1998; 90: 398–399.
Cebrián A, Herrera-Pombo JL, Díez JJ, et al. Genetic and clinical analysis in 10 Spanish patients with multiple endocrine neoplasia type 1. Eur J Hum Genet 1999; 7: 585–589.
Morelli A, Falchetti A, Martineti V, et al. MEN1 gene mutation analysis in Italian patients with multiple endocrine neoplasia type 1. Eur J Endocrinol 2000; 142: 131–137.
Tahara H, Imanishi Y, Yamada T, et al. Rare somatic inactivation of the multiple endocrine neoplasia type 1 gene in secondary hyperparathyroidism of uremia. J Clin Endocrinol Metab 2000; 85: 4113–4117.
Guo SS, Sawicki MP . Molecular and genetic mechanisms of tumorigenesis in multiple endocrine neoplasia type-1. Mol Endocrinol 2001; 15: 1653–1664.
Pannett AA, Thakker RV . Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. J Clin Endocrinol Metab 2001; 86: 4371–4374.
Sato M, Matsubara S, Murao K, Ishida T, Takahara J . Multiple endocrine neoplasia type 1: usefulness of genetic tests in atypical cases. Intern Med 2001; 40: 461–462.
Turner JJ, Leotlela PD, Pannett AA, et al. Frequent occurrence of an intron 4 mutation in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2002; 87: 2688–2693.
Wautot V, Vercherat C, Lespinasse J, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat 2002; 20: 35–47.
Park JH, Kim IJ, Kang HC, et al. Germline mutations of the MEN1 gene in Korean families with multiple endocrine neoplasia type 1 (MEN1) or MEN1-related disorders. Clin Genet 2003; 64: 48–53.
Agarwal SK, Lee Burns A, Sukhodolets KE, et al. Molecular pathology of the MEN1 gene. Ann NY Acad Sci 2004; 1014: 189–198.
Raghavan R, Shah S, Kondkar AA, et al. MEN1 935–1G>C splicing mutation in an Indian patient with multiple endocrine neoplasia type 1. Mol Diagn Ther 2007; 11: 129–131.
Lemos MC, Harding B, Shalet SM, Thakker RV . A novel MEN1 intronic mutation associated with multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2007; 66: 709–713.
Drori-Herishanu L, Horvath A, Nesterova M, et al. An Intronic Mutation is Associated with Prolactinoma in a Young Boy, Decreased Penetrance in his Large Family, and Variable Effects on MEN1 mRNA and Protein. Horm Metab Res 2009; 41: 630–634.
Ellard S, Hattersley AT, Brewer CM, Vaidya B . Detection of an MEN1 gene mutation depends on clinical features and supports current referral criteria for diagnostic molecular genetic testing. Clin Endocrinol (Oxf) 2005; 62: 169–175.
Klein RD, Salih S, Bessoni J, Bale AE . Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 2005; 7: 131–138.
Odou MF, Cardot-Bauters C, Vantyghem MC, et al. Contribution of genetic analysis in screening for MEN1 among patients with sporadic disease and one or more typical manifestation. Ann Endocrinol (Paris) 2006; 67: 581–587.
Jäger AC, Friis-Hansen L, Hansen TV, et al. Characteristics of the Danish families with multiple endocrine neoplasia type 1. Mol Cell Endocrinol 2006; 249: 123–132.
Kishi M, Tsukada T, Shimizu S, et al. A large germline deletion of the MEN1 gene in a family with multiple endocrine neoplasia type 1. Jpn J Cancer Res 1998; 89: 1–5.
Bergman L, Teh B, Cardinal J, et al. Identification of MEN1 gene mutations in families with MEN 1 and related disorders. Br J Cancer 2000; 83: 1009–1014.
Cavaco BM, Domingues R, Bacelar MC, et al. Mutational analysis of Portuguese families with multiple endocrine neoplasia type 1 reveals large germline deletions. Clin Endocrinol (Oxf) 2002; 56: 465–473.
Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjöld M . Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab 2008; 92: 3389–3395.
Fukuuchi A, Nagamura Y, Yaguchi H, Ohkura N, Obara T, Tsukada T . A whole MEN1 gene deletion flanked by Alu repeats in a family with multiple endocrine neoplasia type 1. Jpn J Clin Oncol 2006; 36: 739–744.
Marini F, Carbonell Sala S, Falchetti A, Caramelli D, Brandi ML . The genetic ascertainment of multiple endocrine neoplasia type 1 syndrome by ancient DNA analysis. J Endocrinol Invest 2008; 31: 905–909.
Vasen HF, Lamers CB, Lips CJ . Screening for multiple endocrine neoplasia syndrome type I. Arch Int Med 1989; 149: 2717–2722.
Kytölä S, Villablanca A, Ebeling T, et al. Founder effect in multiple endocrine neoplasia type 1 (MEN 1) in Finland. J Med Genet 2001; 38: 185–189.
Burgess JR, David R, Greenaway TM, Parameswaran V, Shepherd JJ . Osteoporosis in multiple endocrine neoplasia type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Arch Surg 1999; 134: 1119–1123.
Kouvaraki MA, Shapiro SE, Cote GJ, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg 2006; 30: 643–653.
Toledo RA, Lourenco DM, Coutinho FL, et al. Novel MEN1 germline mutations in Brazilian families with multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2007; 67: 377–384.
Lourenço DM Jr, Toledo RA, Coutinho FL, et al. The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (Sao Paulo) 2007; 62: 465–476.
WHO. Technical report series 843. Geneva: World Health Organization, 1994; 5–48.
Lourenço DM Jr, Toledo RA, Mackowiak II, et al. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur J Endocrinol 2008; 159: 259–274.
Falchetti A, Brandi ML . Multiple endocrine neoplasia type I variants and phenocopies: more than a nosological issue?. J Clin Endocrinol Metab 2009; 94: 1518–1520.
Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006; 103: 15558–15563.
Agarwal SK, Mateo CM, Marx SJ . Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 2009; 94: 1826–1834.
La P, Desmond A, Hou Z, Silva AC, Schnepp RW, Hua X . Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene 2006; 25: 3537–3546.
Tala HP, Carvajal CA, González AA, et al. New splicing mutation of MEN1 gene affecting the translocation of menin to the nucleus. J Endocrinol Invest 2006; 29: 888–893.
Sowa H, Kaji H, Hendy GN, et al. Menin is required for bone morphogenetic protein 2- and transforming growth factor β-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem 2004; 279: 40267–40275.
Sowa H, Kaji H, Kitazawa R, et al. Menin inactivation leads to loss of transforming growth factor beta inhibition of parathyroid cell proliferation and parathyroid hormone secretion. Cancer Res 2004; 64: 2222–2228.
Lacerte A, Lee EH, Reynaud R, et al. Activin inhibits pituitary prolactin expression and cell growth though Smads, Pit-1 and menin. Mol Endocrinol 2004; 18: 1558–1569.
Hendy GN, Kaji H, Sowa H, Lebrun JJ, Canaff L . Menin and TGF-beta superfamily member signaling via the Smad pathway in pituitary, parathyroid and osteoblast. Horm Metab Res 2005; 37: 375–379.
Chen YX, Yan J, Keeshan K, et al. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci USA 2006; 103: 1018–1023.
Balogh K, Rácz K, Patócs A, Hunyady L . Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab 2006; 17: 357–364.
Dreijerink KM, Mulder KW, Winkler GS, Höppener JW, Lips CJ, Timmers HT . Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res 2006; 66: 4929–4935.
MacConaill LE, Hughes CM, Rozenblatt-Rosen O, Nannepaga S, Meyerson M . Phosphorylation of the menin tumor suppressor protein on serine 543 and serine 583. Mol Cancer Res 2006; 4: 793–801.
Farley SM, Chen G, Guo S, et al. Menin localizes to chromatin through an ATR-CHK1 mediated pathway after UV-induced DNA damage. J Surg Res 2006; 133: 29–37.
Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL . Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res 2007; 67: 7275–7283.
La P, Yang Y, Karnik SK, et al. Menin-mediated caspase 8 expression in suppressing multiple endocrine neoplasia type 1. J Biol Chem 2007; 282: 31332–31340.
Agarwal SK, Impey S, McWeeney S, et al. Distribution of menin-occupied regions in chromatin specifies a broad role of menin in transcriptional regulation. Neoplasia 2007; 9: 101–107.
Theillaumas A, Blanc M, Couderc C, et al. Relation between menin expression and NF-kappaB activity in an intestinal cell line. Mol Cell Endocrinol 2008; 291: 109–115.
Yokoyama A, Cleary ML . Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008; 14: 36–46.
Marek LR, Kottemann MC, Glazer PM, Bale AE . MEN1 and FANCD2 mediate distinct mechanisms of DNA crosslink repair. DNA Repair (Amst) 2008; 7: 476–486.
Bazzi W, Renon M, Vercherat C, et al. MEN1 missense mutations impair sensitization to apoptosis induced by wild-type menin in endocrine pancreatic tumor cells. Gastroenterology 2008; 135: 1698–1709.
Maillard I, Chen YX, Friedman A, et al. Menin regulates the function of hematopoietic stem cells and lymphoid progenitors. Blood 2009; 113: 1661–1669.
Yan J, Yang Y, Zhang H, et al. Menin interacts with IQGAP1 to enhance intercellular adhesion of beta-cells. Oncogene 2009; 28: 973–982.
Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL . MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA 2005; 102: 14765–14770.
Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA 2005; 102: 14659–14664.
Scacheri PC, Davis S, Odom DT, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet 2006; 2: e51.
Naito J, Kaji H, Sowa H, Hendy GN, Sugimoto T, Chihara K . Menin suppresses osteoblast differentiation by antagonizing the AP-1 factor, JunD. J Biol Chem 2005; 280: 4785–4791.
Bazzi W, Renon M, Vercherat C, et al. MEN1 missense mutations impair sensitization to apoptosis induced by wild-type menin in endocrine pancreatic tumor cells. Gastroenterology 2008; 135: 1698–1709.
Lim LC, Tan MH, Eng C, Teh BT, Rajasoorya RC . Thymic carcinoid in multiple endocrine neoplasia 1: genotype-phenotype correlation and prevention. J Intern Med 2006; 259: 428–432.
Shen HC, He M, Powell A, et al. Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1-directed inactivation of Men1. Cancer Res 2009; 69: 1858–1866.
Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658–5671.
Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al. Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf) 2009; 70: 575–581.
McCallum RW, Parameswaran V, Burgess JR . Multiple endocrine neoplasia type 1 (MEN 1) is associated with an increased prevalence of diabetes mellitus and impaired fasting glucose. Clin Endocrinol (Oxf) 2006; 65: 163–168.
Geerdink EA, Van der Luijt RB, Lips CJ . Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening?. Eur J Endocrinol 2003; 149: 577–582.
Machens A, Schaaf L, Karges W, et al. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 2007; 67: 613–622.
Lairmore TC, Piersall LD, DeBenedetti MK, et al. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type 1 (MEN 1). Ann Surg 2004; 239: 637–645.
Acknowledgements
This review was supported by an unrestricted grant from F. I. R. M. O. Fondazione Raffaella Becagli (to M.L.B.).
Author information
Authors and Affiliations
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Falchetti, A., Marini, F., Luzi, E. et al. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet Med 11, 825–835 (2009). https://doi.org/10.1097/GIM.0b013e3181be5c97
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181be5c97
Keywords
This article is cited by
-
Multiple endocrine neoplasia type 1 involving both the liver and lung: a case report
World Journal of Surgical Oncology (2022)
-
Patients’ perception on the quality of care for multiple endocrine neoplasia disorders in Europe: an online survey from a patient support group
Endocrine (2021)
-
Deletion of Menin in craniofacial osteogenic cells in mice elicits development of mandibular ossifying fibroma
Oncogene (2018)
-
Identification, genetic testing, and management of hereditary melanoma
Cancer and Metastasis Reviews (2017)
-
When should genetic testing be performed in patients with neuroendocrine tumours?
Reviews in Endocrine and Metabolic Disorders (2017)
