
Abstract
Purpose: The Ohio State University was one of the first medical centers to begin routinely performing immunohistochemical staining for the four mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2) on all newly diagnosed patients with colorectal cancer. The results of implementing this testing on a clinical basis are critically assessed.
Methods: From March 1, 2006, to March 31, 2008, 270 newly diagnosed colorectal cancer tumors received immunohistochemical staining for MLH1, MSH2, MSH6, and PMS2. If any stain was absent, the cancer genetic counselors were alerted, so that they could contact the patient. A follow-up genetic consultation was recommended for all patients with any stain absent other than MLH1 and to patients with absence of MLH1 ± PMS2 who were diagnosed younger than 60 years had a multiple Lynch syndrome-associated cancers or had a first-degree relative with colorectal cancer or endometrial cancer. Those attending the genetic consultation were offered appropriate follow-up testing.
Results: There were 57 (21.1%) cases with abnormal immunohistochemical results. Genetics was able to contact 54 (94.7%) of these patients. It was determined that 34 (62.9%) of these 54 patients should be referred for a cancer genetics consultation, however, only nine (26.5%) made an appointment. Seven of the nine underwent additional testing, which was informative in five of the patients. Two (0.7%) new cases of Lynch syndrome were diagnosed and three patients were found to have proven/probable MLH1 promoter methylation.
Conclusions: Routine immunohistochemical of the mismatch repair proteins on all newly diagnosed patients with colorectal cancer can be implemented clinically, however, patient uptake of follow-up genetic consultation is lower than expected.
Similar content being viewed by others
Main
Lynch syndrome (or hereditary nonpolyposis colorectal cancer [CRC]) predisposes individuals to develop CRC and other visceral malignancies that are associated with deleterious germline mutations in the DNA mismatch repair (MMR) genes MLH1, MLH2, MSH6, and PMS2. Lynch syndrome is inherited in an autosomal dominant pattern and is the most common inherited predisposition to CRC occurring in approximately 2% to 7% of all CRCs in the United States.1
Before routine molecular testing, Lynch syndrome was diagnosed using clinical criteria alone.2,3 In practice, the sensitivity and specificity of these criteria for diagnosing Lynch syndrome are poor.4 With the advent of molecular diagnostic techniques, it was determined that Lynch syndrome-related cancers are characterized by microsatellite instability (MSI)5 and the absence of MMR protein expression in associated tumors.6 Current guidelines support the use of clinical criteria to guide which tumors are screened for MSI and/or absence of MMR protein expression.7 This combined clinical–molecular diagnostic strategy improves both the sensitivity and specificity for diagnosing Lynch syndrome.8
However, the majority of data from which the clinical criteria are derived involve populations at high risk for an inherited predisposition to CRC. Recent evidence suggests that when testing unselected colorectal adenocarcinomas from the general population for MSI or lack of MMR protein expression, the average age at time of cancer diagnosis is higher than those used as cutoffs to initiate molecular testing in current guidelines.9,10 This may result in the inadvertent underdiagnosis of Lynch syndrome in the general population.
To avoid this problem, it has been suggested that all CRCs should be screened routinely for an MMR defect. This has been accomplished on a research basis with great success with the identification of 44 patients with Lynch syndrome out of 1566 patients with CRC tested and through further genetic counseling and testing, an additional 109 relatives were diagnosed with Lynch syndrome and 140 relatives were found not to have inherited Lynch syndrome.11,12 However, in the research setting, patients were not charged for the screening test, the genetic counseling, or the genetic testing. In fact, the genetic counselor and nurse would travel to the patients' homes to provide the counseling and blood draws for their convenience. Data are lacking on the practical application of this policy into clinical practice where all of the services will be charged to the patient or their insurance, and the patients will have to travel to the medical center for genetic services. We sought to determine the practicality of diagnosing Lynch syndrome with the prospective evaluation of MMR protein expression in all cases of CRC regardless of age or family history as part of routine clinical practice.
MATERIALS AND METHODS
All cases of primary CRC at a single institution (The Ohio State University [OSU] Medical Center) were prospectively stained to examine the expression of the MMR proteins. Cases from outside institutions, biopsies, or metastatic resections were excluded. Patients were provided with a fact sheet about immunohistochemistry (IHC) staining for the MMR proteins (see Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A85) but were not asked to provide informed consent because this was implemented as part of routine clinical practice. IHC was performed to examine the expression of MLH1, MSH2, MSH6, and PMS2 on formalin-fixed, paraffin-embedded tissue, as described in Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A85 using the following primary mouse monoclonal antibodies: MLH1 (1:40 dilution; BD Biosciences Pharmingen, San Diego, CA), MSH2 (1:200 dilution; Calbiochem, San Diego, CA), MSH6 (1:400 dilution; BD Biosciences Pharmingen), and PMS2 (1:200 dilution; BD Biosciences Pharmingen). Positive and negative controls (normal colonic mucosa) stained appropriately, and any convincing nuclear staining was considered positive.
The stains were read by a gastrointestinal pathologist and protein expression was reported as absent or present. If a tumor exhibited an absence of an MMR protein, the clinical cancer genetics program was informed with previous authorization from the surgeon of record. A genetic counselor then attempted to contact the patient by phone and/or letter to review the IHC result and offer a genetic consultation. Those patients with an absence of MLH1 and PMS2 on IHC were encouraged to schedule a genetic consultation if they were diagnosed younger than 60 years or if they had a positive family history (at least one first-degree relative with colorectal or endometrial cancer). All patients with any other abnormal IHC result were encouraged to schedule a genetic consultation. At the genetic consultation, further testing was discussed and ordered based on indication and patient preference with informed consent. All patients found to have Lynch syndrome and affected family members were then referred for intensive Lynch syndrome cancer surveillance.
RESULTS
From March 1, 2006, through March 31, 2008, 270 cases of primary colorectal were diagnosed at our institution. Of these, 21.1% (57 of 270) stained absent for one or two MMR proteins (Table 1). As expected, the most common abnormal result was an absence of the MLH1 and PMS2 proteins. Thirty-eight (66.7%) tumors had this result. Ten tumors (17.5%) were absent for MSH6 only. Seven (12.3%) tumors were missing both MSH2 and MSH6. One tumor (1.8%) was absent for PMS2 alone. In addition, there was one tumor (1.8%) that lacked expression of both MSH6 and PMS2. Finally, one tumor (1.8%) was missing MLH1, PMS2, and MSH6. IHC stains in the clinical practice are routinely reimbursed at 50% to 60% of the charged cost. Reimbursement for the four MMR protein stains in this study was obtained at a level similar to other IHC stains used in the clinical practice.
Of the 57 individuals whose tumor stained absent for MMR proteins, 54 (94.7%) were successfully contacted by the clinical cancer genetics program thus far. It should be noted, however, that 5 (8.8%) of the patients were deceased, so that results had to be provided to next-of-kin. In addition, 7 (12.3%) of the patients were prisoners who could not be contacted by phone but letters were sent to them in the care of their prison warden, encouraging their family members to attend a genetic consultation. Of the 38 patients with an absence of MLH1 and PMS2, 12 were diagnosed younger than 60 years and 3 of the patients diagnosed at the age of 60 years or older had a positive family history or synchronous or metachronous Lynch syndrome primaries, so we recommended that they receive a cancer genetics consultation. As a result, based on the IHC result and personal and family history, it was determined that 34 of the 54 (63%) patients who were contacted should receive a cancer genetics consultation.
Eighteen individuals (31.6% of all abnormal IHC cases; 52.9% of those for whom counseling was recommended) made an appointment with the Clinical Cancer Genetics Program. Nine individuals (15.8% of all abnormal IHC cases; 26.5% of those for whom counseling was recommended) received a cancer genetic consultation thus far (Table 2); the remaining nine individuals cancelled their appointments with Genetics. Broken down by the result, 1 of the 7 individuals with an absence of MSH2 and MSH6 was seen in Genetics (2 cancelled), 2 of the 10 individuals with an absence of MSH6 alone were seen in Genetics (2 cancelled), 5 of the 46 individuals with an absence of MLH1 and PMS2 were seen in genetics (4 cancelled), and 1 of the 3 patients with other abnormal IHC results was seen in Genetics (1 cancelled).
Of the nine individuals who had a cancer genetic consultation, 7 (77.8%) elected to pursue additional testing. One individual was found to have a methylation of the MLH1 promoter and a somatic BRAF mutation effectively ruling out a diagnosis of Lynch syndrome. Two individuals have been diagnosed with Lynch syndrome (one each with a deleterious mutation in MLH1 and MSH6). Four patients have had additional genetic testing but no mutation has been identified; two of these were patients with an absence of MLH1 and PMS2 with negative sequencing and large rearrangement testing of the MLH1 gene, indicating that the patients likely have methylation of the MLH1 promoter although this testing was not ordered. In a cohort of 270 patients with colon cancer, 7.6 individuals would be expected to have Lynch syndrome because of a germline mutation in an MMR gene (0.028 × 270).11,12 Therefore, on the basis of these previous studies, we believe that we have only identified 26.3% (2 of 7.6) of the expected number of Lynch syndrome cases in this series of patients.
Case 1 was diagnosed with an 8.5-cm moderately differentiated adenocarcinoma of the cecum at the age of 37 years. None of the 45 lymph nodes sampled were involved with cancer. IHC results indicated that the tumor had an absence of the MLH1 and PMS2 proteins (Fig. 1). After a genetic consultation, MLH1 sequencing and large rearrangement testing were ordered through a clinical laboratory. Genetic testing identified a IVS9(790)+1G>A germline mutation in the MLH1 gene and confirmed a diagnosis of Lynch syndrome. His family history (Fig. 2) was remarkable for a father with a small intestinal cancer diagnosed at the age of 46 years and a colon cancer diagnosed at the age of 48 years. His sister had a cervical cancer at the age of 35 years, a paternal uncle was diagnosed with lymphoma in his 40s and died at the age of 55 years, and a paternal aunt was diagnosed with a breast cancer at the age of 53 years. He meets the Bethesda criteria for further evaluation of his tumor for possible Lynch syndrome. Notably, so did his father who was treated for both of his primary cancers at OSU but had never been referred to cancer genetics. This case illustrates that not all cases that should be referred to cancer genetics are referred and can be picked up through routine IHC of all newly diagnosed patients with CRC.

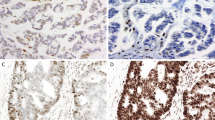
Immunohistochemical staining results for Case 1. There is an absence of the MLH1 and PMS2 proteins and presence of the MSH2 and MSH6 proteins.

Pedigree for Case 1.
Case 40 was diagnosed with a 3.5 × 3.0 × 1.0 cm mucinous adenocarcinoma of the cecum at the age of 53 years. He did not have any of 19 lymph nodes involved with cancer. His tumor IHC results showed an absence of MSH6 and focal staining for MSH2 (Fig. 3). After a genetic consultation, MSH2 sequencing and large rearrangement testing and MSH6 sequencing were ordered through a clinical laboratory. Genetic testing indicated that this patient had an IVS5–2A>G Y germline mutation of the MSH6 gene. His family history (Fig. 4) includes a paternal grandfather with a colon cancer diagnosed at the age of 64 years, a paternal uncle with a lymphoma diagnosed in his 50s, and a paternal first cousin with a breast cancer diagnosed at the age of 48 years. Although this tumor is mucinous, thus meeting the histology component of the Bethesda guidelines, the patient's family history alone does not meet the Bethesda guidelines.

Immunohistochemical staining results for Case 40. There is absence of the MSH6, focal presence of MSH2, and absence of the MLH1 and PMS2 proteins.

Pedigree for Case 40.
DISCUSSION
Our study shows that the routine evaluation of CRC for MMR proteins regardless of age at onset or family history is feasible. Furthermore, we found that the application of this policy is well received by both physicians and genetic counselors. However, uptake of genetic counseling by patients with abnormal IHC results is poor (26.5% of those for whom it is recommended) in the clinical setting. Although it might be expected that compliance with counseling and testing would be lower in the clinical setting than in the research setting (where counseling and testing were free and where we agreed to provide the genetic counseling at patient's home if necessary), this dramatic finding was not anticipated. Patients may not have been as motivated for genetic counseling and/or genetic testing because they did not seek it out themselves. Common barriers identified by the genetic counselors were cost, time, and lack of interest. There was no particular subset of patients who seemed more likely to undergo genetic counseling. Both males and female subjects underwent genetic counseling at similar rates. Furthermore, the majority of patient referrals are from the same geographic area making travel distance irrelevant. In addition, an unanticipated finding was that multiple individuals (12.3%) with abnormal IHC results in our series were prisoners making follow-up extremely challenging. Further, five (8.8%) individuals were deceased by the time genetics attempted to call out their abnormal IHC result although this is occurring less frequently now that genetics is receiving the abnormal results immediately from pathology. We are continuing to consider adjustments to this process to maximize follow-up genetic counseling and testing. The genetic counselors now plan to offer to meet patients with abnormal IHC at one of their already scheduled follow-up appointments with surgery or oncology at OSU. They will provide genetic counseling at that time and order genetic testing if the patient elects to pursue it. Although all patients who pursue genetic testing will receive their results by phone, we will encourage those who are found to have Lynch syndrome to undergo a full outpatient consultation in Genetics during which time they can bring additional family members who may be interested in pursuing testing.
The majority of patients (77.8%) who were counseled elected to pursue additional genetic testing. Of those tested, informative results were obtained for five of seven (three cases with likely/proven MLH1 promoter methylation and two cases of Lynch syndrome). This finding suggests that our approach will detect individuals more likely to have Lynch syndrome and that additional testing is useful for clarifying abnormal IHC results. Furthermore, the cases of Lynch syndrome who are detected through routine screening of all newly diagnosed patients with CRC may have gone undetected otherwise either due to lack of referral to cancer genetics or because the families do not meet any published criteria for the diagnosis of Lynch syndrome.
MLH1 hypermethylation was likely the most common cause of an absence of MMR protein by IHC in our population as it is in the general population.13 In this study, we tried to limit the number of cases with methylation of the MLH1 promoter who received a genetic consultation by screening patients diagnosed at the age of 60 years or older and only referring those with synchronous or metachronous Lynch syndrome primaries or a positive family history (defined as one first-degree relative with colon or endometrial cancer). We have recently implemented BRAF mutation testing as an adjunct to IHC for all cases with an absence of MLH1 and PMS2. We do not plan to contact patients who are found to have a BRAF mutation given that this has nearly 100% specificity for ruling out Lynch syndrome. This should lessen the workload for the genetic counselors who are calling out these results and limit the number of individuals who are alarmed unnecessarily that they may have Lynch syndrome.
A second potential limitation to our study could be the cost incurred by routine IHC screening of all colorectal carcinomas for an absence of MMR proteins. The IHC analysis was covered by insurance providers at a level similar to other IHC stains, minimizing direct cost to the patient. However, further cost-benefit analyses are necessary to further evaluate this process. It is known that most of the benefits are accrued by the testing of at-risk relatives of the probands found to have Lynch syndrome as part of this screening process. With only two new Lynch syndrome families diagnosed after 2 years of clinical screening for Lynch syndrome, the number of at-risk relatives who have benefited from testing is small.
CONCLUSION
IHC staining for MMR proteins is relatively easy to institute in the routine evaluation of CRC. Furthermore, routine testing does not lead to substantial additional testing and is reimbursed at levels similar to other IHC stains. However, patient's uptake of genetic consultation after an abnormal IHC result is lower than expected which will limit our ability to diagnose all patients with Lynch syndrome.
References
Lynch H, de la Chapelle A . Genomic medicine: hereditary colon cancer. N Engl J Med 2003; 348: 919–932.
Vasen H, Mecklin JP, Khan P, Lynch HT . The International Collaborative Group on Hereditary Non-polyposis Colorectal Cancer. Dis Colon Rectum 1991; 34: 424–425.
Vasen H, Watson P, Mecklin JP, Lynch HT . New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999; 116: 1453–1456.
Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE . Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet 2000; 37: 641–645.
Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J . Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997; 57: 4749–4756.
Thibodeau SN, French AJ, Roche PC, et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 1996; 56: 4836–4840.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–268.
Pinol V, Castells A, Andreu M, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005; 293: 1986–1994.
Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 2005; 129: 415–421.
Salovaara R, Loukola A, Kristo P, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000; 18: 2193–2200.
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008; 26: 5783–5788.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851–1860.
Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95: 6870–6875.
Acknowledgements
We thank all the cancer genetic counselors at OSU; Dawn Allain, Rebecca Nagy, Robert Pilarski, Leigha Senter, and Kevin Sweet for their work in contacting the patients with abnormal IHC results. We also thank Victoria Schunemann for her assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
South, C., Yearsley, M., Martin, E. et al. Immunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancer. Genet Med 11, 812–817 (2009). https://doi.org/10.1097/GIM.0b013e3181b99b75
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181b99b75
Keywords
This article is cited by
-
Universal tumor screening for Lynch syndrome: perspectives of Canadian pathologists and genetic counselors
Journal of Community Genetics (2019)
-
Clinical Impact of Mismatch Repair Protein Testing on Outcome of Early Staged Colorectal Carcinomas
Journal of Gastrointestinal Cancer (2018)
-
Rad51C-ATXN7 fusion gene expression in colorectal tumors
Molecular Cancer (2016)
-
Universal Screening of Colorectal Cancers for Lynch Syndrome: Challenges and Opportunities
Digestive Diseases and Sciences (2016)
-
Reflex testing for Lynch syndrome: If we build it, will they come? Lessons learned from the uptake of clinical genetics services by individuals with newly diagnosed colorectal cancer (CRC)
Familial Cancer (2014)
