
Abstract
Purpose: Previous studies of connexin-related hearing loss have typically reported on mixed age groups or adults. To further address epidemiology and natural history of connexin-related hearing loss, we conducted a longitudinal study in an ethnically diverse cohort of infants and toddlers under 3 years of age. Our study compares infants with and without connexin-related hearing loss to examine differences in the prevalence of connexin and non-connexin-related hearing loss by ethnic origin, detection by newborn hearing screening, phenotype, neonatal risk factors, and family history. This is the first study to differentiate infants with and without connexin-related hearing loss.
Methods: We enrolled 95 infants with hearing loss from whom both exons of Cx26 were sequenced and the Cx30 deletion was assayed. Demographic, family history, newborn hearing screening data, perinatal, and audiologic records were analyzed.
Results: Genetic testing identified biallelic Cx26/30 hearing loss-associated variants in 24.7% of infants with a significantly lower prevalence in Hispanic infants (9.1%). Eighty-two infants underwent newborn hearing screening; 12 infants passed, 3 had connexin-related hearing loss. No differences in newborn hearing screening pass rate, neonatal complications, or hearing loss severity were detected between infants with and without connexin-related hearing loss. Family history correlates with connexin-related hearing loss.
Conclusions: Connexin-related hearing loss occurs in one quarter of infants in an ethnically diverse hearing loss population but with a lower prevalence in Hispanic infants. Not all infants with connexin-related hearing loss fail newborn hearing screening. Family history correlates significantly with connexin-related hearing loss. Genetic testing should not be deferred because of newborn complications. These results will have an impact on genetic testing for infant hearing loss.
Similar content being viewed by others

Main
Hearing loss is common and occurs in 1–3 per thousand newborns.1,2 Universal newborn hearing screening (NHS) performed shortly after birth employs bedside physiologic hearing screening modalities, automated auditory brainstem evoked response (ABR), or otoacoustic emissions (OAE), with results, pass or refer (fail) provided to parents and medical providers.3 The rationale behind universal NHS is evidence based. Timely intervention, before 6 months of age, results in better language outcomes. Infants diagnosed in toddler months generally do not achieve communication skills on par with peers.4,5
Hearing loss-associated variants (we have employed the phrase ‘hearing loss-associated variant' to express a change in DNA sequence that when present, leads to the phenotypes of deafness or hard of hearing) in the gene GJB2, encoding the gap junction protein, Cx26, are the most common genetic cause of nonsyndromic prelingual sensorineural hearing loss worldwide.6–9 Cx26 and the genomically and functionally related gene, Cx30, are abundantly expressed in the cochlea.10 In the Midwestern United States, hearing loss-associated variants in Cx26 are identified in nearly 50% of individuals with nonsyndromic sensorineural hearing loss.11
Connexin-related hearing loss is autosomal recessive; to have hearing loss, an individual must inherit two hearing loss-associated variants, one from each parent. Carrier frequencies for hearing loss-associated variants range from 1 to 4%11–13 comparable with the carrier frequency in the white population for cystic fibrosis (3–4%). Rare autosomal dominant Cx26 alleles have also been reported and are frequently syndromic with dermatologic disorders.14 There are common founder alleles identified within various ethnic groups.14,15 35delG is the most common in the white population, 167delT is the most common in the Ashkenazi population, 235delC and V37I predominate in the Asian population, and R135W in the East African population.16 A deletion of 309 kb in Cx30 was identified and is known as del(GJB6-D13S1830). This Cx30 deletion inherited in trans with a Cx26 hearing loss-associated variant results in hearing loss.17,18
Current clinical practice advocates genetic testing for infants diagnosed with hearing loss.19 Discussions have been presented toward inclusion of genetic testing into NHS protocols.20,21 One step toward inclusion of genetic testing into NHS practice requires the characterization of the prevalence of Cx26/30 hearing loss-associated variants, ideally in diverse populations. Data regarding the frequency of connexin-related hearing loss in non-white populations in the United States is largely unknown, particularly in individuals of Mexican American origin residing in Southern California.22
The onset of connexin-related hearing loss begins prelingually with suggested congenital onset.16 This would indicate that NHS would sufficiently identify all infants with connexin-related hearing loss. Recent case reports suggest that some infants with connexin-related hearing loss were not identified by NHS.23–25 Systematic correlation of NHS results and genetic testing in a series of infants has not been carried out.
The phenotype of connexin-related hearing loss is typically described as nonsyndromic and sensorineural, ranging in severity from mild to profound.15,26 Significant genotype/phenotype correlations have been established; protein truncating variants produce severe to profound hearing loss whereas amino acid substitutions produce mild to moderate hearing loss.15 However, comparisons of hearing loss severity between infants with connexin-related hearing loss versus those without connexin-related hearing loss have not been carried out to determine a priori which infants are more likely to have connexin-related hearing loss.
Risk factors associated with infant hearing loss include neonatal intensive care unit stay, aminoglycoside exposure, infections, and hyperbilirubinemia.27 Correlation of neonatal hearing loss risk factors with Cx26/30 gene status has not been performed to determine whether infants who already have hearing loss risk factors would be more or less likely to have connexin-related hearing loss.
We report the results of a study to determine the prevalence of Cx26/30 genotypes and allele frequencies in a diverse US population of infants with hearing loss. We correlated NHS outcomes, hearing loss severity and common neonatal factors such as prematurity, birth weight, Apgar scores, hyperbilirubinemia, aminoglycoside exposure, and family history with presence or absence of hearing loss-associated variants to determine factors to help predict which children are more likely to have connexin-related hearing loss. This is the first study of this kind that makes an effort to differentiate children with connexin-related hearing loss from those who do not.
METHODS
Participant recruitment
Parents of potentially eligible infants and toddlers were made aware of our study by brochures placed in nurseries, clinics, and education centers or through referral from audiology, otolaryngology, or intervention-based programs. A second route was through the newborn nursery at UCLA Medical Center. Families (parents and infant/toddler) were enrolled though an informed consent process approved by the relevant Institutional Review Boards. A board certified genetic counselor provided genetic counseling and informed consent directly in English or Spanish.
Infants and toddlers less than 3 years of age diagnosed with hearing loss (n = 99 infants) were eligible to participate. Newborns referred after failing NHS (n = 6 newborns) were also eligible to participate before diagnostic audiometric evaluation.
Parents were offered genetic testing on their infant from a buccal brush specimen. After completion of laboratory studies, nearly all parents returned in person to receive results and genetic counseling. Prenatal, birth, postnatal, NHS, and audiologic records were collected, reviewed, and abstracted throughout the 4-year course of the study. Some or all records were received for 102 of 105 infants and toddlers in the study. Of the 105 infants, 95 were confirmed to have permanent hearing loss and are reported here.
A 3-generation pedigree was obtained with careful attention to history of childhood hearing loss in first degree (parents, siblings), second degree (grandparents, aunts/uncles), and third degree (first cousins) relatives. Demographic information and ethnic and racial identification were obtained from the parents for their infants.
Connexin variant analysis
Genomic DNA was isolated from fresh buccal brush specimens from the participating infants and toddlers using standard reagents (Qiagen, Valencia, CA).
All Cx26/30 diagnostic assays were carried out under standard clinical molecular genetics protocols in the CLIA-certified Orphan Disease Testing Center and Diagnostic Molecular Pathology Laboratory at UCLA.
Variant analysis for the Cx26 gene was carried out by bidirectional sequencing of both exons. Genomic DNA was amplified using primers to exon 1 (Forward: gtgtggggtgcggttaaaaggcgccacgg and Reverse: agggaccgcgagacccagagcggttgc) and exon 2 (Forward: tttcctgtgttgtgtgcattcgt and Reverse: gccagtttaacgcattgccc [bold denotes mismatch]). Two additional internal sequencing primers for exon 2 were designed against genomic sequence (Forward: tcaagggggagataaagagt and Reverse: gaggagatcaaaacccagaag). Amplicons were subjected to dideoxy-sequencing and analysis on either on a Beckmann CEQ 8000 (Beckman-Coulter, Fullerton, CA) or an ABI 3130 (Applied Biosystems, Foster City, CA) capillary electrophoresis system.
The 309kb deletion in the Cx30 gene, del(GJB6-D13S1830), was identified by a published assay.18
Cx26 V27I polymorphism studies
One hundred purified DNA samples from the control population “The Mexican American Population of Southern California” were purchased from the Coriell Institute (Camden, NJ) consisting of genomic DNA from healthy persons having at least three of four grandparents from Mexico. Samples were assayed for the V27I polymorphism by amplification of exon 2 of the gene encoding Cx26 using primers CX26Ex2F (gttcctgtgttgtgtgcattcgt) and CX26Ex2R (gggcaatgcgttaaactggc) and subjected to digestion with BstF51 (New England Biolabs, Ipswich, MA).
Audiometric analysis
For each subject, audiology records were scrutinized to determine degree, configuration, and type of hearing loss in each ear. Thresholds were extracted initially from an electrophysiologic measure, either ABR thresholds (tonal responses if available or clicks if no frequency specific data were available) or in some cases from Auditory Steady State Responses. Thresholds obtained by behavioral evaluation such as visually reinforced audiometry or standard audiometric techniques were extracted on older children. In most cases, an ear-specific behavioral response was eventually obtained but when necessary, interpretation of sound-field presentations for determining better ear thresholds was used. Both air and bone conduction pure tone and speech thresholds were evaluated to determine type of hearing loss, when bone conduction was available. In some cases, normal tympanometry was the only indication of the status of the conductive mechanism and if so, this information was used. The OAE evaluations were used to judge test consistency and to determine whether auditory neuropathy was a possible factor.
For most infants, it was possible to compute ear-specific pure tone averages (PTAs) from at least two of four frequencies (500, 1000, 2000, and 4000 Hz) and PTA from the better performing ear was analyzed. Audiometric data from the most recent and reliable evaluation was selected for most analyses. However, multiple audiometric time points were available on many of the infants/toddlers, allowing us to determine type, laterality, and progression of hearing loss. Permanent, sensorineural hearing loss was defined as a combination of absent OAEs, air-bone gap less than 15 dB or at least one normal tympanogram. A conductive component was defined when an air-bone gap greater than 15 dB was present or when multiple abnormal tympanograms were noted. Auditory neuropathy was defined when a subject demonstrated more than a mild hearing loss, poor or absent ABR, and present OAEs. For descriptive purposes, hearing loss severity was defined based on the PTA in decibel hearing loss as ≤20 dB for normal hearing, 21–39 dB for mild hearing loss, 40–69 dB for moderate hearing loss, 70–94 dB for severe hearing loss, >95 dB profound hearing loss.
Statistical analysis
Data were double-entered to minimize error and analyzed using SAS v.9.28 Descriptive statistics were computed for detection of anomalies, examination of variable distributions, and descriptive purposes. Due to non-normality of the distributions of PTAs, gestational age, Apgar scores and birth weight, comparison of these variables as a function of number of hearing loss-associated variants was analyzed using the nonparametric Kruskal-Wallis test. The proportion of heterozygotes in our sample was compared with the expected population proportion using a test of a single proportion. The frequency of the V27I allele in our sample was compared with the frequency of the V27I allele in a control sample using the test of two proportions. Fisher exact (FE) test was used to test for independence between number of hearing loss-associated alleles and ethnic origin, frequency of newborn complications, and family history of hearing loss and between NHS result and screening method. Statistical significance was set at 0.05. For analysis of patients with Cx26/30 hearing loss-associated variants, both truncating and nontruncating were grouped together. Variants listed in Table 2 are “hearing loss-associated variants” and not to be confused with polymorphic variants such as V27I.
RESULTS
Over a three and one half year period, December 2002 through June 2006, infants and toddlers were enrolled into the study. Six newborns referred for outpatient NHS entered the study before diagnostic audiometric evaluation. Of these infants, five passed outpatient NHS and one infant had moderate unilateral sensorineural hearing loss. Ninety-nine infants and toddlers entered the study after parental report of a diagnosis of hearing loss. Diagnosis of hearing loss could not be confirmed in three infants because of unavailability of audiology records and two infants were determined to be hearing.
The final analysis group consisted of 95 infants with confirmed permanent hearing loss. Two sets of siblings are included in this sample for a total of 93 independent families. Table 1 provides descriptive information on this sample.
The race and ethnicity of the infants of this cohort was as follows: Hispanic origin (37%) with bi-parental origins primarily from Mexico (n = 21). The mean age at diagnosis for 88 infants for whom the initial diagnostic audiology records were available was 7.4 months (SD = 8.3). The mean age of enrollment was 13.2 months (SD = 10.8) (Table 1).
Genotypes consistent with autosomal recessive connexin-related hearing loss were identified in 23 families yielding a frequency of 24.7% (93 genetically independent infants). Genotypes are listed in Table 2.
In 10 infants, only one hearing loss-associated variant was identified, yielding a heterozygote frequency of 10.8% in the sample of 93 independent infants. This proportion of infants carrying only one hearing loss-associated variant is significantly greater than the reported United States carrier frequency of 3.01% (P < 0.001).11 For 64.5% of the infants in this study, no hearing loss-associated variants were identified (Table 2).
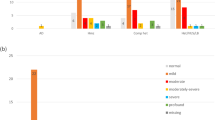
We found connexin-related hearing loss in 33.3% of infants of non-Hispanic white origin, in 46.2% of infants of Asian origin, and 9.1% in infants of Hispanic, primarily Mexican, origin. The frequency of connexin-related hearing loss in infants of Hispanic origin is significantly lower than that found in infants of non-Hispanic white and Asian origin (FE, P = 0.04), suggesting that biallelic changes in Cx26/30 are less frequently observed as the cause of hearing loss in infants of Hispanic origin (Fig. 1).

Hearing loss-associated variant detection rate by ethnicity. These data exclude the second sibling with hearing loss in the two families with two participating infants. **Stars denote a statistically significantly lower frequency of biallelic hearing loss variants in persons of Hispanic origin compared to non-Hispanic white and Asian infants. Infants of African American heritage and mixed heritage were excluded from this analysis.
In infants of Hispanic origin, 21 of whom were of Mexican origin, a high frequency of the Cx26 V27I allele not in association with any other allele was observed (supplemental Table 1, available online only). To determine whether this allele is associated with hearing loss we tested 100 control samples. We found that the frequency of V27I in our sample of infants of Mexican origin was 19% and not significantly different than the prevalence in control samples at 24% (P = 0.6). V27I is a polymorphic variant and not associated with hearing loss.29
In this study, we calculated the allele frequency for the hearing loss-associated variants identified over the group of 93 independent infants. As expected, 35delG was the most commonly encountered allele in this population with a frequency of 0.13 of total possible alleles including “wildtype,” followed by V37I (0.05), and 167delT (0.03) (supplemental Table 2, available online only). Other alleles, 235delC, del(GJB6-D13S1830), V27I;E114G (in cis)30 and M34T were each present at a frequency of 0.01. All other alleles were present at a frequency of <0.01. The distribution of alleles between ethnic groups showed differences. The 35delG was highly prevalent in infants of non-Hispanic white origin (allele frequency = 0.25) but also prevalent in patients of Hispanic origin (0.08). The allele 167delT was found in both infants of non-Hispanic white and Hispanic origin (0.08 and 0.02, respectively). The alleles 235delC, V37I, and V27I;E114G (in cis) were only found in infants of Asian origin (supplemental Table 2, available online only).
In this study, 82 infants underwent bedside NHS (Table 3). Information on screening method was available on 69 of these infants: 64 of 69 (92.8%) were screened with ABR, four (5.8%) with OAE, and one (1.5%) with a combination of ABR and OAE. When stratified by screening result, screening method data were available on 87% of those who failed NHS and on 67% of those who passed NHS. There was no statistical association between screening result and use of ABR or OAE (FE, P = 0.36). There was no statistical association between screening result and number of connexin-related hearing loss associated variants (FE, P = 0.38).
Seventy of the 82 screened infants failed NHS, with a mean age at hearing loss diagnosis of 4.9 months (SD = 5.8). Twenty infants, 29% of those identified by NHS, were identified to have connexin-related hearing loss. Thus, connexin-related hearing loss occurs in a significant proportion of infants who fail NHS.
Twelve infants with hearing loss passed NHS (Table 4). In this group, five had some hearing thresholds in the mild range in at least one ear. Of these five infants not detected by the NHS process, one was found to be homozygous for the V37I Cx26 hearing loss-associated variant. This infant was screened by ABR and his hearing loss was diagnosed at about 21 months of age. The V37I variant is the result of a missense mutation and data suggest that this variant produces a mild to moderate range hearing loss.15,31,32 Hence, infants with this Cx26 variant may be more likely to pass NHS as a result of a milder audiologic phenotype than infants with protein truncating variants.
Seven of the 12 infants who passed the NHS were found to demonstrate bilateral severe to profound hearing loss. The chance of passing the screening in both ears for an infant with severe, bilateral hearing loss is very small. Of these seven, two (unrelated) infants were known to display biallelic Cx26 variants with one having an older sibling with hearing loss. In both cases, the infants were compound heterozygotes for 35delG and 167delT. One infant, with an older sibling with hearing loss, was screened with OAE and was identified at approximately 2 months of age with profound hearing loss. The other infant was screened with ABR and was identified about approximately 1 year of age with a severe hearing loss. As there was no evidence of hearing loss progression (data available upon request), that is, no change in hearing levels over time once detected, in these infants, we must consider the possibility that this genotype predisposes to delayed onset of hearing loss. Our data suggests that nonpenetrance of connexin-related hearing loss at birth is 8%.
Table 4 shows the types of hearing loss in our sample separated by genotype. For infants with connexin-related hearing loss, all infants had bilateral sensorineural hearing loss with severity ranging from mild to profound (Tables 4 and 5). For infants who did not have connexin-related hearing loss, the hearing loss phenotypes were more variable and included unilateral, mixed, conductive, and auditory neuropathy. These data show that infants without connexin-related hearing loss have a broader range of phenotypes than infants with connexin-related hearing loss. The difference in PTA as a function of number of hearing loss-associated variants among the 85 infants with bilateral sensorineural hearing loss was significant (Kruskal-Wallis F(2,82) = 3.56, P = 0.03). The median PTA (Table 5) from infants without connexin-related hearing loss was more severe.
We examined the rates of neonatal risk factors as a function of genotype (supplemental Table 3, available online only). These analyses focused on infants with either two or zero connexin-related hearing loss-associated variants. We found no statistical difference in gestational age or birth weight or differences in Apgar scores, aminoglycoside exposure, hyperbilirubinemia, sepsis, or meningitis between groups. Our sample size provided 70–100% power to detect differences on the quantitative variables as small as 0.67 SD, and 70% power to detect a difference between proportions of at least 20%.
We studied the association of family history with number of allelic variants (Table 6). Family history of hearing loss has been listed as one of the risk factors for hearing loss.27 Although nearly all infants in this study were born to hearing parents, through obtaining a three-generation pedigree on each participating family, we identified 17 families with a history of childhood hearing loss in at least one family member. Taken in aggregate, the presence of either first, second, or third degree relatives in the family history was significantly correlated with the presence of biallelic variants in Cx26/30 (FE, P = 0.003). Thus, presence of childhood hearing loss in a family member increased the likelihood of connexin-related hearing loss caused by biallelic hearing loss-associated variants in Cx26/30 (odds ratio = 6.8, 95% confidence interval = [2.37–17.7]). In the scenario of a positive family history, notably a second degree relative with hearing loss, the index of suspicion for connexin-related hearing loss should be increased.
DISCUSSION
We analyzed data from a longitudinal study to determine prevalence of connexin-related hearing loss in an ethnically diverse group of infants and toddlers under 3 years of age confirmed to have hearing loss and correlated the prevalence of hearing loss-associated variants with ethnic origin, NHS status, hearing loss severity, family history, and newborn risk factors.
Participants entered the study via parental interest from brochures placed in nurseries, clinics, and education centers or from recommendations of our study from audiologists, otolaryngologists, and deaf educators. In this way, our sample may be biased toward infants with more severe hearing loss or parents who were more motivated to participate in our study.
In this diverse group of infants, ∼25% were confirmed to have connexin-related hearing loss. This prevalence falls within the range of previously published prevalence of Cx26/30 hearing loss-associated variants of 12–16% in a predominantly pediatric age group of mixed hearing loss type33–35 to 40–50% in populations of adults and children with apparently nonsyndromic sensorineural hearing loss.6,11 However, as our study group was relatively homogenous in the age of ascertainment and type of hearing loss but diverse in ethnic representation, the prevalence of connexin-related hearing loss differed predominantly by ethnicity and family history.
Patients of Hispanic origin and non-Hispanic whites were nearly equally represented in our study population, (37% and 36%, respectively). Participation of families of Asian origin, 14%, was comparable to the American Community Survey.36 However, few participants of African American origin entered the study (1%) when compared with the Los Angeles demographic of 7.8%.
Our genetic testing strategy revealed hearing loss-associated variants in ∼33% of white infants and ∼46% of Asian infants. However, there was a surprisingly low rate of connexin-related hearing loss, ∼9%, in Hispanic infants. The low rates of connexin-related hearing loss in a cohort of Hispanic infants primarily of Mexican, Central and South American origin has not been previously tabulated but has been implied in other studies.29,35 Low rates of connexin-related hearing loss have been tabulated in populations of African American and Caribbean Hispanic origin in New York City.37 This suggests there are other causes contributing to hearing loss in this predominantly Mexican and Central American Hispanic population requiring further study.
Data from the “Hispanic Health and Nutrition Examination Survey: 1982–1984” showed the prevalence of hearing loss >15 dB in children of Mexican American heritage to be 27.6 per 1000 compared with non-Hispanic white children with a prevalence of 15.5 per 1000 suggesting that the prevalence of hearing loss among Hispanic children is more common than non-Hispanic white children. Despite the low prevalence of connexin-related hearing loss among children of Hispanic heritage, there is a higher rate of hearing loss. This observation suggests a pressing need to identify the major causes of hearing loss in the Mexican American population as they will be underserved by any effort to include genetic testing into NHS protocols.
For 10.8% of the infants with hearing loss, only a single hearing loss-associated variant was identified. This observed frequency of single alleles is greater than the observed carrier frequency of 3.01% in the US Midwestern population.11 It is very unlikely that the single alleles could be dominant, as the identified sequence variants have been published previously as recessive and because in our patients we had no indication of a clear autosomal dominant family history in any of the subjects. Additionally, two of the alleles, S129N and Y152X, are rarely identified,15 suggesting that they are unlikely to be polymorphisms and have a role in the pathogenesis of hearing loss. Other groups identified a higher than expected number of single hearing loss variants.38 Thus, the presence of a single recessive allele in an individual with hearing loss may not simply represent a carrier state and should be interpreted cautiously when providing genetic counseling. Other factors, such as infection, may be co-occurring with mutations in Cx26/3039 warranting further investigation.
We calculated the allele frequency for hearing loss-associated variants and found seven alleles at a frequency of >1% and these included 35delG, V37I, 167delT, 235delC, del(GJB6-D13S1830), V27I;E114G (in cis), and M34T. Skewing of allele prevalence by ethnicity was observed and these observations will be important for development of genetic testing panels, much like those developed for cystic fibrosis.40
In this study, we identified 12 infants with hearing loss who passed NHS. Screening results were not associated with number of connexin-related hearing loss associated variants or with screening methods in this sample, as both ABR and OAE revealed pass results in some cases, and so other reasons should be explored to explain this pass rate. It is not unexpected that five infants with mild/moderate hearing loss within this group were not identified by NHS. Data from Norton et al.41 demonstrated that fail rates for all basic types of hearing screening including ABR, and distortion product or transient OAEs, do not approach 100% until the degree of hearing loss reaches or exceeds 40 dB. In other words, infants with any hearing thresholds of 40 dB or better may be expected to pass the NHS. As one of the infants in this group was a V37I homozygote, we must assume that the degree of hearing loss was better than 40 dB shortly after birth. Additionally, within this group of 12 infants, three had connexin-related hearing loss and had hearing loss in the severe to profound range. Not all infants with connexin-related hearing loss may be detected by bedside NHS and for some infants, the onset of severe hearing loss may be later than in the immediate newborn period or so mild in some frequency regions that they would go undetected. We estimate the rate of nonpenetrance of connexin-related hearing loss to be 8% at birth. These data collected in a systematic manner support previous case based reports that some infants with connexin-related hearing loss will pass NHS.23–25,42
In this study, we compared hearing loss severity between groups of infants with and without hearing loss-associated variants. In infants with connexin-related hearing loss, SNHL was the only phenotype. There were significant differences between groups of infants on the basis of number of hearing loss associated variants, with those who had connexin-related hearing loss in aggregate showing better PTA than those without. However, given the spread of phenotypes associated with truncating and missense variants,15 we must caution that severity of hearing loss alone cannot be used to predict which infant should be offered genetic testing for Cx26/30 although the type of hearing loss, SNHL, would be more predictive of connexin-related hearing loss.
We wanted to determine whether the rate of neonatal complications differed as a function of number of Cx26/30 hearing loss-associated variants. We evaluated neonatal risk factors in our population and compared them with the presence or absence of hearing loss-associated variants. Neonatal risk factors were observed in infants with and without connexin hearing loss-associated variants. However, we found no significant difference between groups as a function of the number of alleles and neonatal factors such as birth weight, gestational age, Apgar scores, hyperbilirubinemia, sepsis, meningitis, and aminoglycoside exposure. We conclude that genetic testing for hearing loss should not be deferred based on the presence of neonatal complications. Additional studies with larger samples and additional factors are warranted to examine associations between hearing loss genotypes and neonatal risk factors.
One factor that significantly correlates with connexin-related hearing loss is the presence of other family members with hearing loss, most notably, second degree relatives such as grandparents and parental siblings. We found that the presence of a positive family history increased the odds of having connexin-related hearing loss approximately 7-fold and contrasts strongly with the infants who did not have connexin-related hearing loss. Although this seems intuitively obvious, in nonconsanguineous families, the proband with the recessive condition is frequently the first person in the family with the condition. Again, we must caution that absence of a family history should not preclude genetic testing for hearing loss, however, a family history of hearing loss increases suspicion.
In summary, connexin-related hearing loss can be identified in nearly a quarter of infants with hearing loss from a diverse US population residing in Southern California. Biallelic hearing loss-associated variants confirming the diagnosis of autosomal recessive connexin-related hearing loss are more prevalent in infants of non-Hispanic white and Asian origin (33.3% and 46.2%, respectively) with a significantly lower rate among infants of Hispanic origin (9%). Identification of a single hearing loss-associated variant is common in infants with hearing loss (10.8%) and significantly greater than the established US carrier frequency of 3% strongly suggesting that identification of a single hearing loss-associated variant in an individual with hearing loss should be interpreted with caution and may not simply represent a carrier state. Although most infants with connexin-related hearing loss will be identified by NHS, a nontrivial subset of genetically predisposed infants will pass screening and remain at risk for hearing loss. Family history of childhood hearing loss increases suspicion that an infant will have connexin-related hearing loss. The presence of neonatal complications does not preclude the possibility that an infant will have connexin-related hearing loss.
References
Finitzo T, Albright K, O'Neal J . The newborn with hearing loss: detection in the nursery. Pediatrics 1998; 102: 1452–1460.
Fortnum HM, Summerfield AQ, Marshall DH, et al. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ 2001; 323: 536–540.
Centers for Disease Control. Early Hearing Detection and Intervention (EHDI) Program. 2007. Available at: http://www.cdc.gov/NCBDDD/ehdi/default.htm. Accessed June 5, 2008.
Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL . Language of early-and later-identified children with hearing loss. Pediatrics 1998; 102: 1161–1171.
Moeller MP . Early intervention and language development in children who are deaf and hard of hearing. Pediatrics 2000; 106: e43.
Denoyelle F, Weil D, Maw MA, et al. Prelingual deafness: high prevalence of a 30delG mutation in the Connexin 26 gene. Hum Mol Genet 1997; 6: 2173–2177.
Estivill X, Fortina P, Surrey S, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998; 351: 394–398.
Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997; 387: 80–83.
Prasad S, Cucci RA, Green GE, Smith RJ . Genetic testing for hereditary hearing loss: Connexin 26 (GJB2) allele variants and two novel deafness-causing mutations (R32C and 645-648delTAGA). Hum Mutat 2000; 16: 502–508.
Rabionet R, Gasparini P, Estivill X . Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mutat 2000; 16: 190–202.
Green GE, Scott DA, McDonald JM, et al. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA 1999; 281: 2211–2216.
Abe S, Usami S, Shinkawa H, et al. Prevalent Connexin 26 gene (GJB2) mutations in Japanese. J Med Genet 2000; 37: 41–43.
Morell RJ, Kim HJ, Hood LJ, et al. Mutations in the Connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 1998; 339: 1500–1505.
Estivill X, Gasparini P . The Connexin-deafness homepage. Available at: http://davinci.crg.es/deafness Accessed June 4, 2008.
Snoeckx RL, Huygen PL, Feldmann D, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 2005; 77: 945–957.
Kenneson A, Van Naarden Braun K, Boyle C . GJB2 (Connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med 2002; 4: 258–274.
Pallares-Ruiz N, Blanchet P, Mondain M, et al. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect?. Eur J Hum Genet 2002; 10: 72–76.
del Castillo I, Villamar M, Moreno-Pelayo MA, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002; 346: 243–249.
American College of Medical Genetics Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genetic evaluation of congenital hearing loss expert panel. ACMG statement. Genet Med 2002; 4: 162–171.
Morton CC, Nance WE . Newborn hearing screening–a silent revolution. N Engl J Med 2006; 354: 2151–2164.
Schimmenti LA, Martinez A, Fox M, et al. Genetic testing as part of the early hearing detection and intervention (EHDI) process. Genet Med 2004; 6: 521–525.
Kenneson A, Myers MF, Lubin IM, Boyle C . Genetic laboratory practices related to testing of the GJB2 (Connexin-26) gene in the United States in 1999 and 2000. Genet Test 2003; 7: 49–56.
Norris VW, Arnos KS, Hanks WD, et al. Does universal newborn hearing screening identify all children with GJB2 (Connexin 26) deafness? Penetrance of GJB2 deafness. Ear Hear 2006; 27: 732–741.
Pagarkar W, Bitner-Glindzicz M, Knight J, Sirimanna T . Late postnatal onset of hearing loss due to GJB2 mutations. Int J Pediatr Otorhinolaryngol 2006; 70: 1119–1124.
Ramsebner R, Volker R, Lucas T, et al. High incidence of GJB2 mutations during screening of newborns for hearing loss in Austria. Ear Hear 2007; 28: 298–301.
Cohn ES, Kelley PM, Fowler TW, et al. Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 1999; 103: 546–550.
Joint Committee on Infant Hearing Year 2007 position statement: principles and guidelines for early hearing detection and intervention programs. Pediatrics 2007; 120: 898–921.
SAS Version 9, SAS Institute: Cary NC.
Tang HY, Fang P, Ward PA, et al. DNA sequence analysis of GJB2, encoding Connexin 26: observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A 2006; 140: 2401–2415.
Pandya A, Arnos KS, Xia XJ, et al. Frequency and distribution of GJB2 (Connexin 26) and GJB6 (Connexin 30) mutations in a large North American repository of deaf probands. Genet Med 2003; 5: 295–303.
Cryns K, Orzan E, Murgia A, et al. A genotype-phenotype correlation for GJB2 (Connexin 26) deafness. J Med Genet 2004; 41: 147–154.
Huculak C, Bruyere H, Nelson TN, et al. V37I Connexin 26 allele in patients with sensorineural hearing loss: evidence of its pathogenicity. Am J Med Genet A 2006; 140: 2394–2400.
Dent KM, Kenneson A, Palumbos JC, et al. Methodology of a multistate study of congenital hearing loss: preliminary data from Utah newborn screening. Am J Med Genet 2004; 125C: 28–34.
Yaeger D, McCallum J, Lewis K, et al. Outcomes of clinical examination and genetic testing of 500 individuals with hearing loss evaluated through a genetics of hearing loss clinic. Am J Med Genet A 2006; 140: 827–836.
Putcha GV, Bejjani BA, Bleoo S, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med 2007; 9: 413–426.
United State Census Bureau. American Community Survey 2005. Available at: http://factfinder.census.gov. Accessed August 5, 2007.
Samanich J, Lowes C, Burk R, et al. Mutations in GJB2, GJB6, and mitochondrial DNA are rare in African American and Caribbean Hispanic individuals with hearing impairment. Am J Med Genet A 2007; 143: 830–838.
Kimberling WJ . Estimation of the frequency of occult mutations for an autosomal recessive disease in the presence of genetic heterogeneity: application to genetic hearing loss disorders. Hum Mutat 2005; 26: 462–470.
Ross SA, Novak Z, Kumbla RA, et al. GJB2 and GJB6 Mutations in Children with Congenital Cytomegalovirus Infection. Pediatr Res 2007; 61: 687–691.
Grody WW . Cystic fibrosis: molecular diagnosis, population screening, and public policy. Arch Pathol Lab Med 1999; 123: 1041–1046.
Norton SJ, Gorga MP, Widen JE, et al. Identification of neonatal hearing impairment: transient evoked otoacoustic emissions during the perinatal period. Ear Hear 2000; 21: 425–442.
Green GE, Smith RJ, Bent JP, Cohn ES . Genetic testing to identify deaf newborns. JAMA 2000; 284: 1245
Acknowledgements
Funding for this project came primarily from R01-DC0055663, with contributions from the University of Minnesota Undergraduate Research Opportunities Program (UROP) and the University of Minnesota, Department of Pediatrics. We express our deep gratitude to the families who participated in our study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Schimmenti, L., Martinez, A., Telatar, M. et al. Infant hearing loss and connexin testing in a diverse population. Genet Med 10, 517–524 (2008). https://doi.org/10.1097/GIM.0b013e31817708fa
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31817708fa
Keywords
This article is cited by
-
The high frequency of GJB2 gene mutation c.313_326del14 suggests its possible origin in ancestors of Lithuanian population
BMC Genetics (2016)
-
Suggestion of GLYAT gene underlying variation of bone size and body lean mass as revealed by a bivariate genome-wide association study
Human Genetics (2013)
-
A systematic review and meta-analysis of 235delC mutation of GJB2 gene
Journal of Translational Medicine (2012)
