
Abstract
Purpose: This study reviews prenatal findings in two cases with a suspected diagnosis of Cornelia de Lange Syndrome, a multisystem disorder characterized by somatic defects and mental retardation, that were later confirmed by postmortem examination and molecular testing. Although the correlation between the Cornelia de Lange Syndrome genotype and phenotype is still unclear, preliminary data indicate several severe phenotypic features that are likely to be detected prenatally in NIPBL-mutated patients.
Methods: We report on two prenatal/neonatal cases with unusual pathologic findings indicating Cornelia de Lange Syndrome. The first, with suspected Cornelia de Lange Syndrome after a set of typical dysmorphisms was noted by prenatal ultrasound, was confirmed by a physical examination after termination of the pregnancy. The second was diagnosed neonatally on the basis of typical clinical signs. Medical complications led to death within the first month of life.
Results: Molecular analysis of NIPBL, the gene that codes for delangin (a component of the cohesin complex), performed postnatally detected two de novo mutations: a missense change (P2056L) in a highly conserved residue and a nonsense alteration (S2490 replaced by a stop codon).
Conclusion: We suggest that early diagnosis of Cornelia de Lange Syndrome would be made much easier by the assemblage of a set of prenatal diagnostic features and criteria in Cornelia de Lange Syndrome cases that have been confirmed by direct physical and molecular examinations. We also suggest that Cornelia de Lange Syndrome genotype–phenotype correlations need to be extended to prenatal cases.
Similar content being viewed by others

Main
Cornelia de Lange Syndrome (CdLS) (Mendelian Inheritance in Man 122470 and Mendelian Inheritance in Man 300590) is a multisystem genetic disorder characterized by prenatal and postnatal growth retardation, typical facial features, upper limb malformations, and delayed psychomotor development. It is a well-delineated, although variable, condition, and its frequency may be as high as 1 in 10,000 live births. The syndrome is usually sporadic, but rare familial occurrences have been reported in approximately 1% of cases.1
Craniofacial features include microbrachycephaly, arched and long eyebrows with synophrys, long eyelashes, low-set ears, small upturned nose, long philtrum, thin lips, downturned corners of the mouth, and microretrognathia. Upper limb malformations range from severe ulnar reduction defects to almost normal limbs. The lower extremities are less involved.
In 2004, mutations of the NIPBL gene located at 5p13 and coding for the protein delangin associated with the cohesion complex2 were identified in some sporadic and familial cases. Molecular screening identified NIPBL frameshift, nonsense, splice site, and missense mutations in approximately 50% of individuals.3–8
A second locus for CdLS was recently identified. Mutations in the X-linked SMC1L1 gene, which encodes a subunit of the cohesin complex, were found in three affected male members of the same family and in one sporadic case.9
Prenatal suspicion of CdLS has rarely been reported,10–17 and the correct diagnosis is almost always made retrospectively. Recently, a fetus with interstitial del5(p13.1p14.2) diagnosed prenatally because of abnormal sonographic findings was confirmed at birth to have CdLS, resulting from complete deletion of NIPBL.18 Suspicious ultrasound findings are usually the identification of upper limb defects, often asymmetric and unilateral, the presence of a congenital diaphragmatic hernia, visceral anomalies, and symmetric intrauterine growth retardation (IUGR), which is present in 95% of cases and becomes more obvious after the 25th week of gestation.16 Over time, published reports have added other ultrasonographic signs, such as an abnormal nuchal translucency.14,19 Prenatal recognition of dysmorphic features became possible with the use of the latest generation of ultrasound equipment that enabled measurement of the precise morphometric parameters of the fetal face, but this technique requires specific and considerable experience.14,20,21
We report on the combined pathologic–genetic investigation of two CdLS cases. The first one was suspected because of typical dysmorphisms on ultrasound and was confirmed by a physical examination after termination of the pregnancy. The second one was diagnosed neonatally on the basis of a typical facial dysmorphism, congenital malformations, and severe growth retardation. Medical complications led to death within the first month of life. In both cases a molecular analysis of NIPBL performed postnatally confirmed the clinical diagnosis.
CASES AND METHODS
Case 1
A 40-year-old primigravida (G1P0) with an unremarkable family history presented for routine ultrasound examination at the 21st week. There was no consanguinity or history of exposure to teratogens. Cytogenetic analysis of amniotic fluid cells previously performed for maternal age had revealed a normal male karyotype (46, XY). To increase understanding of the fetal condition, repeated ultrasound scans were performed and demonstrated the following: at the 20th + 6 week the biparietal diameter was 5 cm (50th centile), the head circumference was 17.3 cm (25th centile), the femur length was 3.2 cm (25th–50th centile), and the abdominal circumference was 16.1 cm (50th centile). Further evaluation of morphology was difficult because of the fetal position. At the 22nd week, dilatation of the vena cava, increased density of the right pulmonary basis, possible thickening of the right ventricular wall, and a hyperechogenic right heart were noted. A second-level ultrasound was carried out at the 22nd + 1 week. The results were as follows: biparietal diameter 5.7 cm (75th centile), head circumference 20.2 cm (50th centile), femur length 3.6 cm (25th–50th centile), and abdominal circumference 18 cm (50th–75th centile). Facial dysmorphisms included a mild micrognathia and a prominent filtrum, and mild cephalic edema was detected. Clinodactyly of the fifth finger and mid-phalanx hypoplasia were also present. An anechogenic area compatible with esophageal duplication was described, together with mild chest hypoplasia as measured by transverse thoracic curve and chest circumference. Echocardiography revealed a hydropericardium and mild cardiac hypertrophy. A dysmorphism of the profile of the cranial bones was identified, as was frontal bossing, mild hypertelorism as calculated from standard referral growth curves of the ocular region, micrognathia, and a prominent philtrum. (Fig. 1A and B). Edema of subcutaneous tissues and an undefined esophageal anomaly were also suggested, as previously reported.

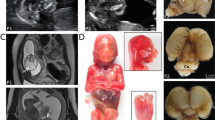
Case 1: (A and B) Frontal view and profile of fetus at 22 weeks (three-dimensional ultrasound). Note frontal bossing, mild hypertelorism, prominent long philtrum, and micrognathia. (C and D) Sagittal and axial single-shot fast spin-echo T2-weighted sections from prenatal magnetic resonance imaging study showing frontal bossing (black arrow) and subcutaneous edema and thickening (asterisk). (E and F) Fetal features at postmortem examination. Evidence of peculiar craniofacial dysmorphisms: synophrys, small nose with anteverted nostrils, smooth and prominent philtrum, thin lips, microretrognathia, and low-set posteriorly rotated ears. (G) Close-up of fetal hand: brachydactyly and clinodactyly of the fifth finger. (H) Gross view of supernumerary spleens in the gastrosplenic ligament (white triangles) and histologic section of ectopic splenic tissue (white arrow) in pancreatic parenchyma (black arrow).
The central nervous system anatomy was apparently normal at ultrasound scan, but fetal magnetic resonance imaging was performed to increase the chance of detecting subtle cortical anomalies. The examination revealed only that the biometric parameters of the hemispheres were at the lower limits for gestational age and that there was subcutaneous edema of the nuchal region (Fig. 1C and D).
In view of the uncertainty of the fetal prognosis and the consistent suspicion of a syndromic condition such as CdLS, the parents elected to terminate the pregnancy.
A postmortem pathologic evaluation revealed a 23-week-old fetus with a weight of 580 g (25th–50th centile), crown-heel length of 29 cm (25th centile), head circumference of 20.5 cm (50th centile), and foot length of 3.8 cm. The following craniofacial dysmorphisms were present: synophrys; hypertelorism; small nose with anteverted nostrils; long, prominent, and smooth philtrum; microretrognathia; thin lips; and low-set, posteriorly rotated ears (Fig. 1E and F). Long eyelashes and hirsutism were apparent, as well as brachydactyly and clinodactyly of the fifth finger (Fig. 1G). Multiple organ malformations were also observed, including a trilobated left lung, thymus hypoplasia, Meckel diverticulum, spleen ectopia with five small supernumerary spleens in the gastrosplenic ligament (Fig. 1H), and an anteriorly rotated left renal pelvis. Histology of the pancreas showed ectopic splenic tissue in the pancreatic parenchyma (Fig. 1H). The heart defects and esophageal duplication were not confirmed.
Case 2
A 24-year-old woman (G2P0) was referred to our unit at the 38th week (36 weeks at ultrasound) for delivery. An ultrasound scan, performed at the 15th + 5 week, had detected fetal growth compatible with the 13th + 5 week that was interpreted as late conception. Female genitalia were diagnosed at the 17th week and confirmed in further scans. Fetal biometry was close to the 40th centile until the 34th week when a symmetric intrauterine growth restriction was detected.
Family history was unremarkable with no consanguinity or history of radiation exposure or drugs. The delivery was uneventful, and the APGAR score was 7–8.
At birth, the weight was 1560 g (<3rd centile), length was 38 cm (<3rd centile), and head circumference was 28 cm (<3rd centile). Hypotonia was prominent. Facial dysmorphisms included synophrys, anteverted nostrils, long and smooth philtrum, thin lips, downturned corners of the mouth, and microretrognathia (Fig. 2A). Hirsutism was present, and a bilateral single transverse palmar crease and postaxial hexadactyly of the left hand were detected (Fig. 2B). Echocardiography revealed tetralogy of Fallot (wide subaortic ventricular septal defect with right–left shunt and multiple accessory ventricular septal defect, overriding aorta, aortic valve dysplasia, and severe stenosis and pulmonary atresia). Cryptorchidism and micropenis were reported.

Case 2: (A) Frontal view of newborn. Peculiar facial dysmorphisms: hirsutism; synophrys, anteverted nostrils, long and smooth philtrum, thin lips, microretrognathia, and downturned corners of the mouth. (B) Close-up of left hand: postaxial hexadactyly. (C and D) Sagittal T1-weighted and axial T2-weighted magnetic resonance image showing brachycephaly, inferior cerebellar vermis hypoplasia (white arrow), thinning of corpus callosum (white arrowhead), and reduction of cerebral hemispheres volume, especially at frontal lobes level. Subcutaneous edema is also visible (asterisks). An accidental finding of small intraventricular hemorrhage can be seen.
Further studies included an electroencephalogram showing immature features, a cerebral ultrasound showing poor circumvolutions, and magnetic resonance imaging showing brachycephaly, inferior cerebellar vermis hypoplasia, thinning of the corpus callosum, and reduction of cerebral hemispheric volume, especially at the level of the frontal lobes. Subcutaneous edema was also visible (Fig. 2C and D).
Cytogenetic analysis of peripheral blood revealed a male karyotype (46, XY).
The infant died after 3 weeks.
The overall clinical and instrumental investigations of the two cases are summarized in Table 1. Despite the fact that the two pregnancies were referred at different gestational ages and investigated using different instrumental tools, the index cases displayed as common denominators facial dysmorphisms and hirsutism typical of CdLS, as well as nuchal edema and reduced volume of the cerebral hemispheres. Several other features, such as polysplenia, polydactyly, and heterotopia of spleen, appeared unique because they were rarely reported in previous descriptions of this condition.
Molecular methods
Genomic DNA was extracted by a commercial kit (Promega, Madison, WI), and 59 fragments covering the entire coding sequence of NIPBL were amplified by intronic primers flanking the splice-site exon/intron junctions from exons 2 to 46. Primer sequences and polymerase chain reaction conditions are available on request. Sequence variants were detected by denaturing high-performance liquid chromatography analysis carried out at three different temperatures on the Transgenomic Wave DNA Fragment Analyser (Transgenomic Inc., San Jose, CA). Each heteroduplex conformer was subjected to direct sequencing (ABI 3100 Automated Sequencer, Applied Biosystems, Foster City, CA).
The Clustal X (Strasbourg, France) program was used to evaluate the conservation of the observed sequence changes, whereas chromosomes from 260 control samples were tested to establish the pathogenetic nature of the missense mutation.
RESULTS
NIPBL mutational analysis
Clinical suspicion of CdLS raised by the overall pathologic evaluation of the terminated fetus (case 1) and the deceased baby (case 2) prompted us to screen DNA from the fetal tissue (chorionic villi sampling) of case 1 and the peripheral blood from case 2 for NIPBL mutations, with the entire coding region of NIPBL screened by denaturing high-performance liquid chromatography. In both cases, abnormal conformers were detected and sequenced.
A 6167C>T change in exon 35 leading to a proline-leucine substitution (P2056L) was identified in case 1. This NIPBL sequence alteration, not previously described, arose de novo, being absent in both parents (Fig. 3A), and its polymorphic nature was excluded by screening NIBPL from 260 normal controls. The affected P2056 residue is in a highly conserved region located within the H3 and H4 units of the HEAT domain of delangin (Fig. 3C). A similar mutational analysis of DNA from case 2 identified a 7469C>G sequence change producing the S2490X stop codon that predicts delangin truncation at residue 2490. Consistent with the clear pathogenetic nature of the observed alteration, NIPBL in both parents was normal (Fig. 3B).

Pedigree of cases 1 (A) and 2 (B). Electropherograms from the fetus (A) and the newborn (B) with suspected CdLS diagnosis show the C6167T change in exon 35 of NIPBL gene leading to substitution of the 2056 proline residue with leucine and the C7968G change in exon 44 predicting a protein truncated at the 2490 residue. The electropherograms with the normal sequence in the parents of both cases are also shown. (C) Schematic genomic structure of NIPBL gene with exons (vertical bars) and introns (connecting diagonal lines with the localization of both mutations). The evolutionary conservation of amino acid residue P2056 (framed in red) affected by the missense mutation (lower panel). The multiple alignments of the protein stretch where the affected amino acid residue is embedded attest to the high amino acid sequence homology from Homo sapiens back to Mus musculus, Danio rerio, Drosophila melanogaster, and Caenorhabditis elegans.
DISCUSSION
The pathology and genetic data underline an emerging issue in the diagnosis of rare Mendelian syndromes presenting with typical clinical signs for which the responsible genes have only recently been identified. This concerns the shift of diagnostic suspicion from postnatal to antenatal life, based on the combination of clinical and imaging data pathognomonic for that syndromic condition and its possible confirmation by targeted molecular analysis. Thus, the prenatal finding of CdLS reported by Hulinsky et al.18 has not been validated by molecular analysis, which leaves one of the most consistent clinical–pathologic diagnoses still unresolved. It is likely that the integrated expertise of ultrasonographers, obstetricians, pediatricians, and clinical geneticists will markedly improve the likelihood of accurate clinical diagnoses, which will, in turn, encourage more specific molecular and molecular-cytogenetic testing.
Despite the “a posteriori” nature of our two described cases, which are not yet sufficient to demonstrate the technical feasibility of genetic testing for CdLS within the time constraints essential for a prenatal diagnosis, they do provide the starting point for assembling a set of diagnostic features and criteria that should increase our ability to diagnose these rare genetic diseases early in pregnancy. The impact of such specific diagnoses of rare diseases on genetic counseling and on the choices offered to the family cannot be overemphasized.
The genotype/phenotype correlation in CdLS is unsettled, but so far the consensus is for an association of a milder phenotype with missense mutations as attested to by the prevalence of these kinds of mutations in familial cases.5 However, the position of the affected amino acid residue in delangin and its possible involvement in crucial functional interactions may lead to different phenotypic outcomes. To add further complexity, varying phenotypic presentations of patients bearing the same missense mutation have also been reported,5 suggesting the influence of NIPBL modifiers and possible epigenetic or environmental effects.5
Prenatal diagnosis of CdLS is doubtless even more complicated given the wide variability of the phenotype, so that, for now, CdLS is suspected only when major malformations are present (e.g., skeletal anomalies or diaphragmatic hernia) but is ignored when symptoms are confined to facial dysmorphisms and IUGR. Furthermore, the phenotypic expression evolves during the second and third trimesters and frequently only becomes evident late in pregnancy. Therefore, a meticulous ultrasound evaluation needs to be performed to improve the chances of correctly recognizing CdLS when facial dysmorphisms, IUGR, and suspected malformations are evident during a routine ultrasound examination. It is important to pay special attention to the combination of long eyelids; small nose with anteverted nostril; long, bulging philtrum; thin lips; micrognathia; and low-set ears. According to some authors, even hirsutism should be visible on ultrasound.12,16 Our first case is a good example of minimal prenatal expression, in which the molecular test was crucial to validate the diagnosis. Even in the second case, if we ignore the congenital heart defect and the polydactyly that were actually missed, the baby presented initially only with late IUGR and facial dysmorphisms. The lack of a fetal karyotype made it difficult to assign the correct gender and masked the severe genital malformation that was detected only at birth.
Unfortunately, a mild phenotype cannot predict a good prognosis of intellectual performance; therefore, early diagnosis and the option of termination is one of the aims of the prenatal surveillance even for pregnancies without increased risk.
Mutational screening carried out in patients affected by CdLS demonstrated the occurrence of frameshift, nonsense, missense, intron splicing mutations, and 5′-untranslated region mutations22 at a frequency ranging from 35%6 to 56%,7 and preliminary phenotypic differences between mutation-positive and mutation-negative individuals were identified. The results so far obtained also suggest a trend toward a more severe clinical phenotype in NIPBL-mutated patients and, within this group, a milder phenotype in individuals with missense and 5′-untranslated region mutations compared with other types.
The missense mutation identified in case 1, although of novel description and thus underlying a phenotype not comparable to any other clinically characterized patient, seems to conform to the general rule of missense mutations in both its localization and putative phenotypic effects. A meta-analysis of all known missense mutations8,23 shows a clustering of such mutations in and around the HEAT repeats; indeed, the observed P2056L mutation affects a residue of the delangin sequence between the H3 and H4 units of the HEAT domain. As for the clinical phenotype, although the fetus came to our attention because of nuchal edema and facial dysmorphisms, he lacked upper limb reduction and did not show cardiac or renal anomalies. In addition, case 1 showed no IUGR at 22 gestational weeks. In contrast, severe growth delay and tetralogy of Fallot were present in case 2, who was found to carry a premature stop mutation.
Mutations of this sort are usually associated with a severe phenotype and most likely represent loss of function attributable to nonsense RNA-mediated decay. The novel S2490X mutation identified in case 2 predicts a terminal truncation, but whether the aberrant transcript managed to escape nonsense-mediated decay and gave rise to a truncated protein exerting a transdominant effect is unknown, because transcript analysis was precluded.
The deceased baby did not exhibit limb reduction, a frequent sign in patients with CdLS bearing inactivating mutations, but did show postaxial hexadactyly, an unusual finding in this disease. However, whether the occurrence of this anomaly is a coincidence or may be an effect of the pathogenic mutation is a matter of speculation. Such equivocal findings will be clarified in the future by more complete descriptions of postnatal patients with CdLS who harbor similarly truncating mutations.
Although the NIBPL mutation rate in patients with CdLS is less than 50%, we did find a pathogenic mutation in both of our cases. This result may be related to the strict case selection and evaluation, to chance, or to both. Our two cases, although sharing the typical facial dysmorphisms of CdLS, give further clues about the wide phenotypic spectrum of the syndrome. Furthermore, the role of NIPBL mutations in organ embryogenesis and system patterning will be clarified when additional prenatal cases undergo a detailed pathologic examination.
Confirmation of CdLS by molecular testing in these two cases, whose postmortem evaluation first raised suspicion of the disease, suggests that we ought to reconsider their prenatal signs as the first indications for a prenatal diagnosis of CdLS.
References
Russell KL, Ming JE, Patel K, Jukofsky L, et al. Dominant paternal transmission of Cornelia de Lange syndrome: a new case and review of 25 previously reported familial recurrences. Am J Med Genet 2001; 104 ( 4): 267–276. Review.
Dorsett D . Roles of the sister chromatid cohesion apparatus in gene expression, development, and human syndromes. Chromosoma 2006. Epub ahead of print.
Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, et al. NIPBL, encoding a homolog of fungal Scc2- type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 2004; 36 ( 6): 636–641. Epub 2004 May 16.
Krantz ID, McCallum J, DeScipio C, Kaur M, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004; 36( 6): 631–635.
Gillis LA, McCallum J, Kaur M, DeScipio C, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet 2004; 75( 4): 610–623.
Borck G, Redon R, Sanlaville D, Rio M, et al. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet 2004; 41: e128.
Bhuiyan ZA, Klein M, Hammond P, van Haeringen A, et al. Genotype-phenotype correlations of 39 patients with Cornelia De Lange syndrome: the Dutch experience. J Med Genet 2006; 43( 7): 568–575.
Yan J, Saifi GM, Wierzba TH, Withers M, et al. Mutational and genotype-phenotype correlation analyses in 28 Polish patients with Cornelia de Lange syndrome. Am J Med Genet A 2006; 140( 14): 1531–1541.
Musio A, Selicorni A, Focarelli ML, Gervasini C, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 2006; 38( 5): 528–530.
Golsby LM, McNamara MF, Anderson CF, Quinn DL, et al. Case of the day. Brachman-De Lange syndrome. J Ultrasound Med 1995; 14( 4): 325–326.
Manouvrier S, Espinasse M, Vaast P, Boute O, et al. Brachmann-de Lange Syndrome: pre and postnatal findings. Am J Med Genet 1996; 62: 268–273.
Ranzini AC, Day-Salvatore D, Farren-Chavez D, McLean DA, et al. Prenatal diagnosis of de Lange syndrome. J Ultrasound Med 1997; 16( 11): 755–758.
Boog G, Sagot F, Winer N, David A, et al. Brachmann-de Lange syndrome: a cause of early symmetric fetal growth delay. Eur J Obstet Gynecol Reprod Biol 1999; 85( 2): 173–177.
Sekimoto H, Osada H, Kimura H, Kamiyama M, et al. Prenatal findings in Brachmann-de Lange syndrome. Arch Gynecol Obstet 2000; 263( 4): 182–184.
Urban M, Hartung J . Ultrasonographic and clinical appearance of a 22 week old fetus with Brachman de Lange Syndrome. Am J Med Genet 2001; 102: 73–75.
Le Vaillant C, Quere MP, David A, Berlivet M, et al. Prenatal diagnosis of a “minor” form of Brachmann-de Lange syndrome by three-dimensional sonography and three-dimensional computed tomography. Fetal Diagn Ther 2004; 19 ( 2): 155–159. Review.
Price N, Bahra M, Griffin D, Hanna G, et al. Cornelia de Lange Syndrome in association with a balanced reciprocal translocation involving chromosomes 3 and 5. Prenat Diagn 2005; 25( 7): 602–603.
Hulinsky R, Byrne JL, Lowichik A, Viskochil DH . Fetus with interstitial del(5)(p13.1p14.2) diagnosed postnatally with Cornelia de Lange syndrome. Am J Med Genet A 2005; 137( 3): 336–338.
Huang WH, Porto M . Abnormal first-trimester fetal nuchal translucency and Cornelia De Lange syndrome. Obstet Gynecol 2002; 99( 5 Pt 2): 956–958.
Rustico MA, Lalatta F, Righini A, Spaccini L, et al. The role of integrated imaging techniques for prenatal prediction of phenotype in two cases of facial anomalies. Prenat Diagn 2004; 24( 7): 508–512.
Chaoui R, Heling KS . Three-dimensional ultrasound in prenatal diagnosis. Curr Opin Obstet Gynecol 2006; 18( 2): 192–202.
Borck G, Zarhrate M, Cluzeau C, Bal E, et al. Father-to-daughter transmission of Cornelia de Lange syndrome caused by a mutation in the 5′ untranslated region of the NIPBL gene. Hum Mutat 2006; 27( 8): 731–735.
Strachan T . Cornelia de Lange Syndrome and the link between chromosomal function, DNA repair and developmental gene regulation. Curr Opin Genet Dev 2005; 15 ( 3): 258–264. Review.
Acknowledgements
The authors are indebted to the parents of the case patients for donating samples and clinical information. The NIPBL molecular study was supported by a grant from ISS (Istituto Superiore di Sanità) to Istituto Auxologico Italiano (L. L).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Lalatta, F., Russo, S., Gentilin, B. et al. Prenatal/neonatal pathology in two cases of Cornelia de Lange syndrome harboring novel mutations of NIPBL. Genet Med 9, 188–194 (2007). https://doi.org/10.1097/GIM.0b013e31803183dd
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31803183dd
Keywords
This article is cited by
-
Integrating molecular and structural findings: Wnt as a possible actor in shaping cognitive impairment in Cornelia de Lange syndrome
Orphanet Journal of Rare Diseases (2017)
-
A case of third trimester diagnosis of Cornelia de Lange syndrome
Archives of Gynecology and Obstetrics (2011)
