
Abstract
Purpose: We present the results of carrier studies in 33 relatives of the paternal branch of a spinal muscular atrophy patient with homozygous absence of the SMN1 gene.
Methods and Results: Once linkage and quantitative analyses were performed, a number of first-, second- and third-degree relatives were identified as carriers given that they shared the at-risk haplotype and showed one SMN1 copy. In the fourth-degree relatives, linkage analysis demonstrated discordance with the quantitative results because the members with one copy were carriers of the mutation, but in a different haplotype background. We concluded that two independent mutations were present in this branch of the family. Furthermore, the combination of both methods of analysis allowed us to identify carriers with two SMN1 genes in one chromosome and none in the remaining chromosome.
Conclusions: Carrier testing in spinal muscular atrophy should be performed by employing both quantitative and linkage analyses in order to guarantee accurate carrier identification.
Similar content being viewed by others


Main
Spinal muscular atrophy (SMA) is an autosomal recessive disorder caused by homozygous absence of the SMN1 gene in approximately 90% of the cases. All SMA patients have one to four copies of SMN2,1 which is the highly homologous SMN1 copy. However, the presence of SMN2 is unable to prevent the disease. With an incidence of 1/6,000 to 1/10,000 live births, carrier frequency in SMA is around 1/35–1/60.2,3 Relatives who are at risk of being carriers commonly request genetic counseling and carrier and/or prenatal diagnoses by molecular analysis. Routine tests for molecular diagnosis of the SMA patients do not allow detection of carriers of the hemizygous absence of the SMN1 given that the normal copy of the gene masks the deletion. Carrier diagnosis in these cases relies on gene tracking with markers of the SMN genes4 and on quantitative analysis to determine the SMN1 copy number.5
There are two main situations calling for SMA carrier detection. The first is the identification of the at-risk haplotypes in the affected member and possible carriers under study. If the markers differ from those identified in the index case or in the known carriers of the family, carrier exclusion could be established. If the markers are identical, there is a high probability that the individual under study is a carrier. However, this does not rule out the possibility that the index case is a de novo or a mosaic mutation from one of the parents.4,6,7 The second situation is the study of partners of confirmed carrier relatives. These partners run an approximate 1/35–1/60 risk of being carriers. In this situation, linkage analysis is not useful unless a degree of kinship is detected. To this end, the quantitative analysis detects individuals with one SMN1 copy although 4–5% of the carriers may have two SMN1 genes in one chromosome and none in the other. We report an SMA family in which the application of both linkage and quantitative analyses allowed 1) to identify carriers of the mutation responsible for the disease of the index patient; 2) to reveal other relatives carrying a second mutant allele in a different chromosomal background; and 3) to detect carriers with two SMN1 copies in this group of relatives.
PATIENTS AND METHODS
Patients
We analyzed 37 members of a family with an SMA type II patient for carrier diagnosis. Thirty-three were part of the paternal branch (Fig. 1).

Pedigree of the family reported. All living members were tested. Carriers within first-, second- and third-degree relatives are depicted in black. Carriers within fourth- or higher-degree relatives of the second mutant allele are depicted in gray. Individuals with oblique black bars have the second mutation and two SMN1 genes. Individuals with the small gray spheres have three SMN1 genes. All the remaining members tested were negative for the two at-risk haplotypes (the causative and the second) and had two SMN1 genes.
SMA locus analysis
Screening for deletions in the SMN genes was performed as previously reported.5 C272 (Ag1-CA) and C212 marker analyses were undertaken with slight modifications including the use of FAM and NED as fluorescent dyes to label the forward C272 (Ag1-CA) and C212 primers, respectively. Samples were analyzed in an ABI Prism 3100 (Gene Scan software) (Perkin Elmer-Applied Biosystems). C272 (Ag1-CA) and C212 are multi-copy, high informative markers located at the 5′ end of the SMN genes and their use provides a good estimate of the minimal numbers of SMN1 and SMN2 genes per individual. Thus, for carrier analysis, these multi-copy markers are preferable to flanking markers of the SMN locus4.
SMN1 quantitative analysis
We employed a quantitative method previously described1,5. DNA was diluted to a final concentration of 5 ng/μl. Quantification was performed on the LightCycler Instrument by Roche Diagnostics (Basel, Switzerland). Online measurements of PCR products were based on the use of SYBR Green I. The analysis was performed with the Second Derivative Maximum Method of the LightCycler Software. A DNA external standard carrying four SMN1 copies was employed. This DNA was from an individual with homozygous absence of SMN2 and four C272 (Ag1-CA) and C212 alleles. We used 1.2 μl genomic DNA in concentrations of 1.25, 2.5, 5 and 7.5 ng/μl respectively, corresponding to 1, 2, 4, 6 fictive copies of SMN1. These dilutions were used to calculate the linear regression curve. All samples were processed and measured in duplicate and the final result was reported as the mean of the measures.
RESULTS
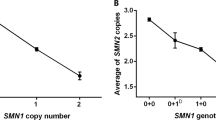
The pedigree illustrated in Figure 1 includes all the individuals tested for linkage and quantitative analyses. Once the at-risk haplotypes were identified, both parents were confirmed as carriers given that they showed one SMN1 copy after quantitative analysis. The patient's mother III-21 inherited a deletion from the maternal grandmother. In the paternal branch, the analysis of a number of relatives showed that seven of them (II-9, II-10, II-11, III-13, III-14, III-18 and III-19) were diagnosed as carriers of the causative mutation of the SMN1 gene given that they shared the at risk haplotype C272 (Ag1-CA) 206–214 and C212 223–225 and had one SMN1 copy (Fig. 2). On the other hand, seven members (II-8, II-12, III-11, III-12, III-15, III-16, III-17) were excluded as carriers because they had a different haplotype and had two SMN1 copies. Initial linkage studies in the fourth degree relatives showed that none of them were carriers of the at risk haplotype identified in the family. However, a quantitative SMN1 analysis revealed one SMN1 copy in individuals II-1, II-2, II-3, II-5, II-7 and III-1, III-2, III-5, III-9 and III-10. Subsequent linkage analyses demonstrated that all these members with one SMN1 copy shared the same haplotype C272 (Ag1-CA) 208–216 and C212 217–219. This second haplotype differed from the at risk haplotype identified in this family branch (Fig. 2). Furthermore, individuals III-7 and III-8 showed the second haplotype despite having two SMN1 copies. The investigation of parent II-4 indicated that he had three SMN1 copies. His son III-6 had also three SMN1 copies and inherited a maternal haplotype, which differed from that of his two brothers. This finding confirmed that III-7 and III-8 were carriers of two SMN1 genes in one chromosome and of none in the other.

Electropherogram of members IV-1 (index case), III-20 (father) and II-5 (paternal aunt) showing the results of the C212 marker. Note the at-risk alleles (223 and 225 peaks) transmitted by the father to the patient and the different alleles (217–219 peaks) associated with the second mutant allele in the paternal aunt. m = ROX molecular weight marker.
DISCUSSION
In this family, we encountered two main shortcomings of SMA carrier diagnoses. The first problem concerns the possibility of incorrect carrier exclusion by analyzing only the C272 (Ag1-CA) and C212 markers, underscoring the limitations of linkage analyses. Individuals II-1, II-2, II-3, II-5, II-7 and III-1, III-2, III-5, III-9 and III-10 (Fig. 1) would have been diagnosed as non-carriers since they did not inherit the corresponding at risk haplotype identified in this branch of the family. However, the quantitative test showed one SMN1 copy. Moreover, the linkage analysis revealed a common haplotype in these individuals. We concluded that a second SMN mutant allele was present in this part of the family. The second problem refers to the limitation of quantitative analysis in discriminating carriers with two SMN1 genes. We focused on individuals III-7 and III-8, whose mother II-5 showed one SMN1 gene. Her sons, despite inheriting the common at-risk haplotype shared by this part of the family, showed two SMN1 genes in repeated quantitative tests. By contrast, in their brother III-6 we detected three SMN1 genes and the non-risk haplotype inherited from the mother. Finally, the quantitative analysis of their father II-4 also demonstrated three SMN1 genes. Overall, these results indicated that individuals III-7 and III-8 were carriers of the second mutation represented by no SMN1 allele inherited from their mother and two SMN1 genes in cis in the chromosome inherited from their father.
The results of this family illustrate the advantage of using the combination of linkage and quantitative methodologies to overcome the limitations of each analysis applied separately. This has recently been pointed out by Eggermann et al. who described a grandmother with a possible somatic mosaicism for a deletion in the SMN1 gene based on borderline quantitative tests and on the inheritance of three different haplotypes in her offspring.7 Our observation confirms that carrier diagnosis in SMA relatives should be performed using both methodologies. The linkage analysis identifies the alleles that segregate together with the mutation, helping to exclude possible carriers. In those who share the at risk haplotype, the quantitative methods should confirm the presence of one SMN1 gene or should identify the unusual cases with two SMN1 genes in one chromosome. Unrelated partners with one SMN1 copy must be considered as carriers. When partners show a double copy result, they could still run a low residual risk (around 1/900 to 1/1,250) of being a carrier.5,8 Furthermore, the carriers of subtle mutations are not detected with this methodology. To minimize the residual risk, mutation screening can be performed to detect rare subtle mutations i.e., the c.399_402delAGAG in the Spanish population9 or the p.Y272C in the German population.10 Moreover, quantitative studies could allow the identification of new mutations such as the one reported in this family. Given the genetic instability of the SMN region and given the high frequency of carriers in the population, it is not surprising that large family branches analyzed by quantitative and linkage methodologies, as presented here, could yield evidence of second independent mutations.
References
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Pearn J . Classification of spinal muscular atrophies. Lancet 1980; 26: 919–922.
Cusin V, Clermont O, Gerard B, Chantereau D, et al. Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 2003; 40: e39.
Melki J, Lefebvre S, Burglen L, Burlet P, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 1994; 3: 1474–1477.
Cusco I, Barcelo MJ, Baiget M, Tizzano EF . Implementation of SMA carrier testing in genetic laboratories: comparison of two methods for quantifying the SMN1 gene. Hum Mutat 2002; 20: 452–459.
Wirth B, Schmidt T, Hahnen E, Rudnik-Schoneborn . et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61: 1102–1111.
Eggermann T, Zerres K, Anhuf D, Kotzot D, et al. Somatic mosaicism for a heterozygous deletion of the survival motor neuron (SMN1) gene. Eur J Hum Genet 2005; 13: 309–313.
Ogino S, Wilson RB . Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 2002; 111: 477–500.
Cusco I, Lopez E, Soler-Botija C, Barcelo MJ, et al. A genetic and phenotypic analysis in Spanish spinal muscular atrophy patients with c. 399_402del AGAG, the most frequently found subtle mutation in the SMN1 gene. Hum Mutat 2003; 22: 136–143.
Sun Y, Grimmler M, Schwarzer V, Schoenen F, et al. Molecular and funtional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum. Mutat 2005; 25: 64–71.
Acknowledgements
We are indebted to all the family members for their invaluable collaboration in this study.
Author information
Authors and Affiliations
Additional information
This work was supported by FIS 02-1275 and Red Inergen C03/05.
Rights and permissions
About this article
Cite this article
Barceló, M., Alias, L., Caselles, L. et al. Two independent mutations of the SMN1 gene in the same spinal muscular atrophy family branch: Lessons for carrier diagnosis. Genet Med 8, 259–262 (2006). https://doi.org/10.1097/01.gim.0000214319.99550.10
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.gim.0000214319.99550.10
Keywords
This article is cited by
-
Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling
European Journal of Human Genetics (2018)
