
Abstract
Purpose: We tested the hypothesis that Sotos syndrome (SoS) due to the common deletion is a contiguous gene syndrome incorporating plasma coagulation factor twelve (FXII) deficiency. The relationship between FXII activity and the genotype at a functional polymorphism of the FXII gene was investigated.
Methods: A total of 21 patients including those with the common deletion, smaller deletions, and point mutations, and four control individuals were analyzed. We examined FXII activity in patients and controls, and analyzed their FXII 46C/T genotype using direct DNA sequencing.
Results: Among 10 common deletion patients, seven patients had lower FXII activity with the 46T allele of the FXII gene, whereas three patients had normal FXII activity with the 46C allele. Two patients with smaller deletions, whose FXII gene is not deleted had low FXII activity, but one patient with a smaller deletion had normal FXII. Four point mutation patients and controls all had FXII activities within the normal range.
Conclusion: FXII activity in SoS patients with the common deletion is predominantly determined by the functional polymorphism of the remaining hemizygous FXII allele. Thus, Sotos syndrome is a contiguous gene syndrome incorporating coagulation factor twelve (FXII) deficiency.
Similar content being viewed by others

Main
Sotos syndrome (SoS, OMIM no. 117550), first reported by Sotos et al. in 1964, is characterized by overgrowth, specific craniofacial features, developmental delay, and advanced bone age, sometimes associated with microdeletion of chromosome 5q35.1–8 The causative gene for SoS is NSD1, but about half of the cases, especially of Japanese ethnicity, harbor a deletion encompassing NSD1 rather than a point mutation in this gene. An ≈2-Mb common microdeletion predominates, and we and others have demonstrated that there are low-copy repeats (proximal SoS-PREP and distal SoS-DREP) where the breakpoints for the common deletion cluster.6,9,10 The SoS common deletion results from nonallelic homologous recombination (NAHR) utilizing the flanking LCRs as substrates, suggesting that SoS is a condition resulting from DNA rearrangement due to genomic region-specific architecture, also known as a genomic disorder.6,9–11 When the genomic interval involved in the DNA rearrangement includes multiple genes that are also dosage-sensitive, this is termed a contiguous gene syndrome (CGS) and independent endophenotypes may occur.11–13
We encountered an individual with SoS who had low coagulation factor twelve (FXII; also known as Hageman factor) activity in plasma.14 FXII is a member of the contact factors for blood coagulation such as factor IX, prekallikrein, and high molecular weight kininogen.15 In FXII deficiency, a mild bleeding tendency has been reported,16–18 and others have shown that low levels of FXII increased the risk of thrombosis.19–22 The relationship between FXII activity and risk of coronary artery disease is still unclear.23–25 Overall, the significance of abnormal FXII activities, especially FXII deficiency, remains unknown, but it is generally thought to have minimal clinical consequences.
Several factors influence the levels of FXII activity. Missense or nonsense mutations of FXII lead to its low levels in plasma.26–33 The activity of this factor is also strongly determined by a common polymorphism within the FXII gene (46C/T) (RefSNP rs1801020) (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=1801020); with the 46T allele, the translation efficiency of FXII is reduced.34,35
FXII maps to 5q35.3 ≈106 Kb telomeric to NSD1 and within the common SoS microdeletion region (Fig. 1). Herein, we demonstrate that the FXII activity of individuals with SoS is determined by the 46C/T polymorphism in the context of the type of NSD1 mutation. If deletion is the mutational mechanism, then the hypoactive 46T hemizygous allele of FXII can be further exacerbated and result in deficiency. The complex phenotype of SoS suggests deletion of more than one gene may influence ultimate clinical expression and that this condition is a CGS incorporating FXII deficiency as a feature.

Physical map detailing the position of NSD1 and FXII in 5q35.3. Two microsatellite genetic markers, D5S2111 and D5S469, map to this region. The distance between the 3′ end of NSD1 and the 3′ end of FXII is ≈106 Kb. The low copy repeats that mediate the common deletion, proximal SoS-PREP and distal SoS-DREP copies are shown by open rectangles. Bold lines indicate large genome insert bacterial artificial chromosome (BAC) and P1 artificial chromosome (PAC) clones that were used to determine the deletion size.6 The RP11-933K12 clone contains the entire FXII gene. The genomic intervals for the SoS common deletion and smaller deletions are shown by horizontal straight lines under BAC/PAC clones with dashed line representing the nondeleted portion of the genome.
METHODS
Clinical subjects
We enrolled a total of 21 SoS patients (17 males and four females; median age, 5 years, ranging from 9 months to 29 years old), with deletion of NSD1 comprising 17 cases (Table 1). Of these deletions cases, 10 have a de novo common microdeletion (previously described as deletion “A”6 Cases nos. 1–10), three cases had smaller deletions of almost identical sizes without deleting FXII (nos. 11–13) (Fig. 1),6,14 and the size of the deletion in four individuals (nos. 14–17) was not determined. The remaining four patients with SoS (nos. 18–21, all male; median age 4 years old, ranging from 3 to 29 years) have intragenic mutations of NSD1.3,6,36 Four additional individuals were analyzed, the parents of a “small” deletion case,6 and the parents of a common deletion case.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed on all small deletion SoS cases (nos. 11 [ref 14], 12, 13) to determine if the FXII gene was deleted. Two bacterial artificial chromosome (BAC clones) containing the entire FXII gene were used in this study (RP11-933K12, CTB-22D11; UCSC Genome Browser, May 2004 Assembly, http://genome.ucsc.edu/cgi-bin/hgGateway). Probe DNA was labeled with Spectrum Orange-11-UTP (Vysis, Downers Grove, IL) by nick translation, and after hybridization, the signal was detected using fluorescence photomicroscopy.
Laboratory analysis of FXII
FXII activity (normal range: 50–150%) was evaluated with an automatic analyzer (standard clotting time assay using specific plasma without FXII, and activated partial thromboplastin time).
Genotyping of the FXII 46C/T variant
The FXII 46C/T polymorphism was analyzed by direct sequencing using ABI PRISM BigDye Terminators version 3.1 Cycle Sequencing Kit (PE Applied Biosystems).
After amplifying genomic DNA, the PCR products were sequenced using an ABI3100 autosequencer.
RESULTS
Plasma factor XII activity levels were obtained for all subjects and controls. FXII genotypes at the 46C/T functional polymorphism were determined for all individuals in which DNA was available; 19 out of 21 patients and two out of four controls (Table 1). The distribution of FXII activity as a function of mutational status is shown in Figure 2. With the exception of one reported case,14 no individual in this study had any known complications of bleeding, heart disease, or venous thrombosis.

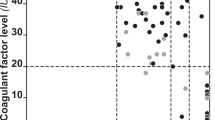
Distribution of FXII activity in plasma for individuals with three different genotypes at the NSD1 locus: heterozygous genomic deletion, wild type, and heterozygous point mutation. Black diamonds (left), white circles (middle), and white squares (right) show FXII activity of deletion patients, parents of deletion patients, and patients with NSD1 intragenic point mutations, respectively. The averages and standard deviations for each group are 49.5 ± 25.5 (deletion), 93.0 ± 22.6 (parents of deletion representing the wild type), and 80.5 ± 14.5 (point mutation).
The mean activity of FXII in SoS individuals with any NSD1 deletion (nos. 1–17) was 49.5% (standard deviation 25.5%) (Table 1). Ten SoS cases had a common microdeletion (nos. 1–10) and of these, seven had low FXII activities, whereas the remaining three cases (nos. 8–10) showed activities within the normal range (86%–107%) (Table 1). As a result of genotyping of the 46C/T functional polymorphism in FXII for the SoS patients with the common deletion, seven patients with low FXII activities had the 46T variant that is associated with reduced FXII activity,34,35 whereas the three SoS patients with normal levels all had the 46C variant on the remaining hemizygous allele (Fig. 3).

Distribution of FXII activity in common deletion individuals with the FXII 46T or 46C polymorphism. The black diamonds indicate the value of FXII activity for individuals with the 46T and 46C polymorphism, respectively. The averages and standard deviations of each group are 38.1 ± 7.8 and 93.7 ± 11.6.
Three SoS cases harbored smaller-sized deletions (nos. 11–13) and FISH analysis was performed to determine if the FXII gene was deleted. In all three instances, the distal breakpoints were localized in the region between BAC clones RP1-118M12 and RP11-933K12. The latter clone containing FXII was not deleted (Fig. 1),3,14 suggesting an intact FXII locus on the deletion chromosome. The assayed FXII activities of these three cases were low except for one (no. 13) who is homozygous for the 46C variant.
Of the four patients with SoS who harbored a point mutation in NSD1 (nos. 18–21), the mean FXII activity was 80.5% (standard deviation 14.5%); an affected patient and his affected father were included in this group (nos. 19 and 20, respectively).36 The FXII activities for four control individuals [the parents of a small deletion patient (nos. 11),14 and of a common deletion patient (nos. 2; Table 1)] were determined and none demonstrated FXII deficiency.
DISCUSSION
In this report, we examined the relationship between FXII activity, a functional polymorphism in the FXII gene, and Sotos syndrome. The majority of the study population were male, but FXII levels are not known to be influenced by age or gender.37 The mechanism of NSD1 disruption among the individuals who were studied was deliberately skewed toward deletion cases, because the primary focus was to correlate the type of the deletion (common type “A” ≈2 Mb microdeletion vs. other types of deletion) with any disruption of the FXII gene, and subsequently, serum FXII activities. The mean FXII activity in all SoS individuals with deletion (including both common and smaller deletions) is statistically lower than that observed for SoS patients with NSD1 point mutation, or for the control parents of SoS children (t test, P = 0.001) (Table 1) (Fig. 2). The FXII activities in the parents (father and mother of patients of nos. 2 and 11) are greater than the activity of their children with deletion (Table 1) (Fig. 2). However, an interesting pattern emerged upon genotyping the 46C/T functional polymorphism of the FXII gene. In SoS patients with the common deletion, the predominant factor influencing FXII activities is the genotype at the hemizygous FXII allele (Fig. 3).
Three of the deletion cases with uncommon smaller deletions (nos. 11–13) were further studied by FISH with probes encompassing the FXII region. In all three, the FXII gene was determined to be present, although two of the cases had FXII deficiency. The FXII activities and genotypes of cases nos. 11, 12, and 13 were 37% (46T/T), 38% (46T/T) and 81% (46C/C), respectively. Thus, the FXII activity was determined by the 46C/T functional polymorphism of the FXII gene similar to the situation in other individuals with two intact alleles.
The FXII activities of the individuals with a deletion of unknown size (nos. 14–17) were either in the deficiency range or borderline low (57%). Unfortunately, the 46C/T functional polymorphism could not be analyzed because DNA was not available (with the exception of nos. 14 and 15). However, due to the Japanese ethnicity and the prevalence of the common microdeletion in this population, they are likely hemizygous for the FXII locus,6 with FXII deficiency arising in a similar fashion as for cases nos. 1 through 7. The FXII activities of the individuals with NSD1 intragenic mutations (nos. 18–21) were all within normal range, demonstrating that the NSD1 intragenic mutation itself does not influence the FXII activities of these SoS patients.
The allele frequency of the 46C/T polymorphisms differs between ethnic populations. In Japan, the T allele (frequency 0.653) is more common than the C allele (ss4935759), whereas in France, the reverse is true (ss3856582) (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=1801020). The ethnic composition of our study population was predominantly Japanese, and among the common microdeletion cases, the frequency of the T allele (7 out of 10, or 0.7) (Table 1) was as expected. Further experimentation is necessary to determine if the prevalence of FXII deficiency in NSD1 deletion cases of SoS in other ethnic groups is lower than what we report in this study, and reflective of differences in the proportion of that population that have the C versus the T allele.
Our results show that serum FXII activities may be a useful and convenient surrogate genetic marker to screen for the SoS common microdeletion. Other congenital anomaly syndromes also have hematological abnormalities. For example, Noonan syndrome is associated with deficiency of coagulation factor XI.38–40 Reports vary on the prevalence of coagulation problems (from 20–74%) but, consistently, a clinically significant bleeding tendency has been noted. However, in contrast to SoS, the genetic basis for this hematological abnormality is unknown.
Recently, targeted deletion mice of FXII have been generated and analyzed.41 Homozygous knockout mice are deficient in serum activities of this factor, whereas heterozygous knockouts have an intermediate activity. No increased incidence of bleeding tendency or venous thrombosis was observed. Thus, consistent with reports in humans, a clinically significant phenotype may not be associated with FXII deficiency.
Knowledge regarding the genetics of SoS has advanced rapidly since its first characterization 40 years ago. In the past few years, we have identified the etiologic gene (NSD1),1–8 and further delineated the mechanistic basis behind its disruption (point mutation vs. microdeletion due to NAHR utilizing flanking LCRs as homologous recombination substrates).6,9,10
Recently, slight differences in clinical manifestations in SoS have been correlated to the type of NSD1 alteration, with deletion individuals having more heart, brain, and genitourinary abnormalities.42 Incorporating FXII deficiency with SoS, this study provides insight into the underlying genetics behind this phenotypic variability. First, the genomic deletion leads to the absence of the primary etiologic gene responsible for the syndrome. Depending on the size of the deletion and differences in breakpoint location, disruption of neighboring genes in the same region may contribute in part to any observed clinical differences. However, also responsible for the phenotypic variability are functional polymorphisms of the remaining allele in any genes now present in a hemizygous state. This study clearly demonstrates this phenomenon with SoS and the 46C/T polymorphism influencing serum FXII activities (Fig. 3).
This concept of genetic variation of the remaining hemizygous alleles influencing phenotype is similar to well-characterized examples.13 In both Prader-Willi syndrome (PWS) and Angelman syndrome (AS), patients hemizygous for the P gene are usually hypopigmented (deletion vs. UPD cases) and unmasking of a recessive P allele can have a further effect on skin color.43,44 Unmasking of a recessive deafness allele, in the MYO15A gene at the DFNB3 locus, is associated with sensorineural deafness in a Smith-Magenis syndrome (SMS) patient with the common SMS deletion.45,46
We hypothesize that other genes6,9,10 in the SoS common deletion interval in addition to FXII may influence the ultimate clinical phenotype. To examine this hypothesis further, chromosome-engineered animal models of SoS deletion may be useful.47 These experiments, along with further studies of the genomic region deleted in SoS patients, are likely to promote further insights into the specific genes that contribute to the clinical phenotype of SoS.
References
Sotos JF, Dodge PR, Muirhead D, Crawford JD, Talbot NB . Cerebral gigantism in childhood. A syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder. N Engl J Med 1964; 271: 109–116.
Cole TRP, Hughes HE . Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31: 20–32.
Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30: 365–366.
Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Rio M, Clech L, Amiel J, Faivre L, Lyonnet S, Le Merrer M et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003; 40: 436–440.
Kurotaki N, Harada N, Shimokawa O, Miyake N, Kawame H, Uetake K et al. Fifty microdeletions among 112 cases of Sotos syndrome: Low copy repeats possibly mediate the common deletion. Hum Mutat 2003; 22: 378–387.
Türkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, Hesse V et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 2003; 11: 858–865.
Kamimura J, Endo Y, Kurotaki N, Kinoshita A, Miyake N, Shimokawa O et al. Identification of eight novel NSD1 mutations in Sotos syndrome. J Med Genet 2003; 40: e126.
Visser R, Shimokawa O, Harada N, Kinoshita A, Ohta T, Niikawa N et al. Identification of a 3.0-kb major recombination hotspot in patients with Sotos Syndrome who carry a common 1.9-Mb microdeletion. Am J Hum Genet 2005; 76: 52–67.
Kurotaki N, Stankiewicz P, Wakui K, Niikawa N, Lupski JR . Sotos syndrome common deletion is mediated by directly oriented subunits within inverted Sos-REP low-copy repeats. Hum Mol Genet 2005; 14: 535–542.
Lupski JR . Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 1998; 14: 417–422.
Schmickel RD . Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr 1986; 109: 231–241.
Shaffer LG, Ledbetter DH, Lupski JR . Molecular Cytogenetics of Contiguous Gene Syndrome: Mechanisms and Consequences of Gene Dosage Imbalance. The Metabolic and Molecular Basis of Inherited Disease. 8th edition. v. I. New York: The McGraw-Hill Companies, Inc., 2001; 1291–1327.
Shen JJ, Kurotaki N, Patel A, Lupski JR, Brown CW . Low factor XII level in an individual with Sotos syndrome. Pediatr Blood Cancer 2005; 44: 187–189.
Tans G, Rosing J . Structural and functional characterization of factor XII. Semin Thromb Hemost 1987; 13: 1–14.
Haanen C, Hommes F, Benraad H, Morselt G . A case of Hageman-factor deficiency and a method to purify the factor. Thromb Diath Haemorrh 1960; 5: 201–217.
Didisheim P . Hageman factor deficiency (Hageman trait). Case report and review of the literature. Arch Intern Med 1962; 110: 170–177.
Ikkala E, Myllylä G, Nevanlinna HR . Rare congenital coagulation factor defects in Finland. Scand J Haematol 1971; 8: 210–215.
Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Am J Hum Genet 2000; 67: 1452–1459.
Soria JM, Almasy L, Souto JC, Bacq D, Buil A, Faure A et al. A quantitative-trait locus in the human factor XII gene influences both plasma factor XII levels and susceptibility to thrombotic disease. Am J Hum Genet 2002; 70: 567–574.
Tirado I, Soria JM, Mateo J, Oliver A, Souto JC, Santamaria A et al. Association after linkage analysis indicates that homozygosity for the 46C→T polymorphism in the F12 gene is a genetic risk factor for venous thrombosis. Thromb Haemost 2004; 91: 899–904.
Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F . The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: A study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis 2004; 17: 139–143.
Endler G, Mannhalter C, Sunder-Plassmann H, Lalouschek W, Kapiotis S, Exner M et al. Homozygosity for the C→T polymorphism at nucleotide 46 in the 5′ untranslated region of the factor XII gene protects from development of acute coronary syndrome. Br J Haematol 2001; 115: 1007–1009.
Colhoun HM, Zito F, Chan NN, Rubens MB, Fuller JH, Humphries SE . Activated factor XII levels and factor XII 46C>T genotype in relation to coronary artery calcification in patients with type 1 diabetes and healthy subjects. Atherosclerosis 2002; 163: 363–369.
Zito F, Lowe GDO, Rumley A, McMahon AD, Humphries SE, on behalf of the WOSCOPS Study Group. Association of the factor XII 46C>T polymorphism with risk of coronary heart disease (CHD) in the WOSCOPS study. Atherosclerosis 2002; 165: 153–158.
Miyata T, Kawabata S-I, Iwanaga S, Takahashi I, Alving B, Saito H . Coagulation factor XII (Hageman factor) Washington D.C.: Inactive factor XIIa results from Cys-571→Ser substitution. Proc Natl Acad Sci U S A 1989; 86: 8319–8322.
Hovinga JK, Schaller J, Stricker H, Wuillemin WA, Furlan M, Lämmle B . Coagulation factor XII Locarno: The functional defect is caused by the amino acid substitution Arg 353→Pro leading to loss of a kallikrein cleavage site. Blood 1994; 84: 1173–1181.
Schloesser M, Hofferbert S, Bartz U, Lutze G, Lämmle B, Engel W . The novel acceptor splice site mutation 11396(G→A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients. Hum Mol Genet 1995; 4: 1235–1237.
Schloesser M, Zeerleder S, Lutze G, Halbmayer W-M, Hofferbert S, Hinney B et al. Mutations in the human factor XII gene. Blood 1997; 90: 3967–3977.
Kondo S, Tokunaga F, Kawano S, Oono Y, Kumagai S, Koide T . Factor XII Tenri, a novel cross-reacting material negative factor XII deficiency, occurs through a proteasome-mediated degradation. Blood 1999; 93: 4300–4308.
Kanaji T, Kanaji S, Osaki K, Kuroiwa M, Sakaguchi M, Mihara K et al. Identification and characterization of two novel mutations (Q421K and R123P) in congenital factor XII deficiency. Thromb Haemost 2001; 86: 1409–1415.
Wada H, Nishioka J, Kasai Y, Kato-Nakazawa K, Abe Y, Morishita Y . Molecular characterization of coagulation factor XII deficiency in a Japanese family. Thromb Haemost 2003; 90: 59–63.
Ishii K, Oguchi S, Moriki T, Yatabe Y, Takeshita E, Murata M et al. Genetic analyses and expression studies identified a novel mutation (W486C) as a molecular basis of congenital coagulation factor XII deficiency. Blood Coagul Fibrinolysis 2004; 15: 367–373.
Kanaji T, Okamura T, Osaki K, Kuroiwa M, Shimoda K, Hamasaki N et al. A common genetic polymorphism (46 C to T substitution) in the 5′-untranslated region of the coagulation factor XII gene is associated with low translation efficiency and decrease in plasma factor XII level. Blood 1998; 91: 2010–2014.
Ishii K, Oguchi S, Murata M, Mitsuyoshi Y, Takeshita E, Ito D et al. Activated factor XII levels are dependent on factor XII 46 C/T genotypes and factor XII zymogen levels, and are associated with vascular risk factors in patients and healthy subjects. Blood Coagul Fibrinolysis 2000; 11: 277–284.
Höglund P, Kurotaki N, Kytölä S, Miyake N, Somer M, Matsumoto N . Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet 2003; 40: 51–54.
Souto JC, Almasy L, Borrell M, Garí M, Martínez E, Mateo J et al. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation 2000; 101: 1546–51.
Kitchens CS, Alexander JA . Partial deficiency of coagulation factor XI as a newly recognized feature of Noonan syndrome. J Pediatr 1983; 102: 224–227.
Sharland M, Patton MA, Talbot S, Chitolie A, Bevan DH . Coagulation-factor deficiencies and abnormal bleeding in Noonan's syndrome. Lancet 1992; 339: 19–21.
Bertola DR, Carneiro JDA, D'Amico EA, Kim CA, Albano LMJ, Sugayama SMM et al. Hematological findings in Noonan syndrome. Rev Hosp Clin Fac Med Sao Paulo 2003; 58: 5–8.
Pauer H-U, Renné T, Hemmerlein B, Legler T, Fritzlar S, Adham I et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo.. Thromb Haemost 2004; 92: 503–508.
Nagai T, Matsumoto N, Kurotaki N, Harada N, Niikawa N, Ogata T et al. Sotos syndrome and haploinsufficiency of NSD1: clinical features of intragenic mutations and submicroscopic deletions. J Med Genet 2003; 40: 285–289.
Lee ST, Nicholls RD, Bundey S, Laxova R, Musarella M, Spritz RA . Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N Engl J Med 1994; 330: 529–534.
Spritz RA, Bailin T, Nicholls RD, Lee S-T, Park S-K, Mascari MJ . Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet 1997; 71: 57–62.
Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet 2001; 109: 535–541.
Friedman TB, Hinnant JT, Ghosh M, Boger ET, Riazuddin S, Lupski JR et al. DFNB3, spectrum of MYO15A recessive mutant alleles and an emerging genotype-phenotype correlation. Adv Otorhinolaryngol 2002; 61: 124–130.
Walz K, Fonseca P, Lupski JR . Animal models for human contiguous gene syndromes and other genomic disorders. Genet Mol Biol 2004; 27: 305–320.
Acknowledgements
This work was supported in part by grants from National Institute of Neurological Disorders and Stroke, National Institutes of Health (R01NS27042), the National Institute of Child Health and Human Development (NIH) (P01 HD38420), and the Baylor College of Medicine Mental Retardation Research Center (HD 2406407). N.K. is a recipient of grant for Medical Research of Alumni Association of Nagasaki University School of Medicine and Nagasaki Medical Association Research Subsidy. We thank the participating families for their cooperation in this study. We also thank Drs. Pawel Stankiewicz and Mehrdad Khajavi for critical review.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Kurotaki, N., Shen, J., Touyama, M. et al. Phenotypic consequences of genetic variation at hemizygous alleles: Sotos syndrome is a contiguous gene syndrome incorporating coagulation factor twelve (FXII) deficiency. Genet Med 7, 479–483 (2005). https://doi.org/10.1097/01.GIM.0000177419.43309.37
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000177419.43309.37
Keywords
This article is cited by
-
Automated prediction of the clinical impact of structural copy number variations
Scientific Reports (2022)
-
The distribution and impact of common copy-number variation in the genome of the domesticated apple, Malus x domestica Borkh
BMC Genomics (2015)
-
Generation of the Sotos syndrome deletion in mice
Mammalian Genome (2012)
-
Hereditary pancreatitis caused by a double gain-of-function trypsinogen mutation
Human Genetics (2008)
-
Unmasking of a hemizygous WFS1 gene mutation by a chromosome 4p deletion of 8.3 Mb in a patient with Wolf–Hirschhorn syndrome
European Journal of Human Genetics (2007)
