
Abstract
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) is a relatively common, underdiagnosed autosomal-dominant disorder of arteriovenous malformations and telangiectases. DNA testing for hereditary hemorrhagic telangiectasia has recently become available in North America, making presymptomatic screening available to relatives with a positive molecular diagnosis. This now enables practitioners to prevent catastrophic complications of undiagnosed pulmonary and CNS arteriovenous malformations and eliminates the need to radiographically screen all at-risk relatives shown to be unaffected by molecular testing. We review the clinical aspects of hereditary hemorrhagic telangiectasia, describe the indications, benefits, and limitations of molecular diagnostic testing for hereditary hemorrhagic telangiectasia, and provide a molecular genetics summary to facilitate genetic counseling before and after DNA testing for this complex disorder.
Similar content being viewed by others

Main
Hereditary hemorrhagic telangiectasia (HHT) is a multisystem vascular dysplasia characterized by solid organ arteriovenous malformations (AVMs) as well as telangiectases of the dermis and mucous membranes. The reported prevalence of HHT varies but has been found to be more than 1/10,000 in well-studied populations, making it a common genetic disorder.1 It has a wide ethnic and geographic distribution.1–4
The external, visible signs (dermal telangiectases and frequent nose bleeding) often do not manifest until the second or third decade of life. Yet internal AVMs in the brain, spinal cord, and lungs are thought to be largely congenital lesions and may present suddenly and with serious complication soon after birth or at an early age.5–8 If recognized, the underlying AVM is usually treatable.
Presymptomatic molecular diagnosis allows for significantly improved care for individuals at risk for HHT. Because the initial clinical presentation of the disorder can be a catastrophic pulmonary or CNS event,5,8–10 presymptomatic diagnosis for relatives of individuals with HHT offers an opportunity for prevention of serious or lethal complications. Individuals shown to be unaffected can be spared unnecessary and costly medical screening.
DESCRIPTION OF HHT
HHT is a characterized by telangiectases and arteriovenous malformations with direct artery-to-vein connections predisposing to shunting and hemorrhage (Fig. 1). Lesions may be small (telangiectases) or large (AVMs) and are located nonrandomly in the body. Telangiectases are found predominantly in the oral and nasal mucosa, tongue, lips, nose, fingertips, and gastrointestinal (GI) mucosa, predominantly of the stomach and small bowel. AVMs occur mostly in the lungs, central nervous system, upper gastrointestinal tract, and liver. The number and location of telangiectases and AVMs vary widely between individuals and within the same family, suggesting that as yet unidentified epigenetic factors and/or modifying genes have a role in their development.

Diagram of normal blood vessel (top) and telangiectasia/AVM (bottom).
To date, mutations in either one of two genes cause HHT and account for most but not all clinical cases. Mutations can occur in the endoglin (ENG) gene on chromosome 9, giving rise to HHT1, or in the activin receptor like kinase 1 (ALK1) gene on chromosome 12 causing HHT 2. Some families do not map to either of these loci, suggesting at least one more gene causing HHT11 (also unpublished data, 2003).
Diagnostic criteria
HHT is diagnosed in an individual who meets three or more of the following diagnostic criteria.12 The diagnosis is considered possible or suspected when two are present and unlikely when fewer than two are present:
-
Spontaneous, recurrent epistaxis; nocturnal nosebleeds heighten concern for HHT
-
Mucocutaneous telangiectases, especially on tongue, lips, oral cavity, fingers, and nose
-
Internal AVM(s) (pulmonary, cerebral, hepatic, gastrointestinal, spinal)
-
First degree relative with HHT according to these criteria
Physical findings
Telangiectasia
Nosebleeds are the most common, usually the earliest and often the most troublesome aspect of HHT. As many as 95% of affected individuals eventually experience recurrent epistaxis, with a mean age of onset of about 12 years and mean frequency of 18 episodes per month. Approximately one half of diagnosed individuals report having onset by age 10 years and 80% to 90% by age 21 years.13 Nosebleeds are spontaneous or start with minimal provocation and frequently occur at night. Although there seems to be a general tendency for nosebleed frequency and severity to increase with age, this is not uniformly the case. Many patients report no particular change in their nosebleeds over time and some even an improvement.14,15 Although nosebleeds may cause chronic anemia and require transfusion in some patients, many do not have nosebleeds that are frequent or severe enough to result in medical treatment or consultation.
Multiple telangiectases of the hands, face, and oral cavity (Fig. 2) occur in a similar percentage of patients but the age of onset is generally 5 to 30 years later than for epistaxis.14,15 It is common for patients to report having first noticed telangiectases in the decade of their 30s. Telangiectases in these locations are often clinically silent but can be sites of bleeding.

Patient with oral telangiectases.
Telangiectases in the GI tract occur anywhere but most commonly in the stomach and upper duodenum. About 25% of individuals over age 60 will have gastrointestinal bleeding, usually presenting with melena or anemia. Bleeding tends to be slow but persistent and may increase in severity with age.16
Arteriovenous malformations
In contrast to the smaller telangiectases, the symptoms of AVMs are often not secondary to hemorrhage. Symptoms of AVMs most often occur as a result of shunting of blood, thrombosis, or embolus.
AVMs in the lungs are thought to be congenital but may enlarge over time.17 They occur in approximately 30% of individuals with HHT.18,19 They may be asymptomatic for many years and may present insidiously or dramatically with respiratory symptoms such as exercise intolerance, cyanosis or pulmonary hemorrhage, migraine headaches, polycythemia, and clubbing.20–23 However, about 30% to 40% of individuals with pulmonary AVMs (PAVMs) will have a central nervous system presentation with thrombotic and embolic events such as stroke, brain abscess, or transient ischemic attacks in the presence of near normal pulmonary arterial oxygen tension.24 Several adverse events typically occur before the PAVM is identified as the source of the CNS events.25 Pregnant women with untreated PAVMs are at high risk for pulmonary hemorrhage.26
Central nervous system AVMs are thought to be congenital too, and new lesions do not appear to develop after birth. Cerebral AVMs (CAVMs) occur in at least 10% of individuals with HHT18,27 and may present at any age as seizure, headache, stroke, or intracranial hemorrhage.28,29 They may present neonatally, in infancy, and in childhood in otherwise asymptomatic children5 (Fig. 3). Reports to date of CAVMs in HHT are likely to underestimate their frequency and contribution to mortality in HHT as those who have a fatal intracranial hemorrhage as the first symptom are rarely diagnosed as having HHT.

Brain MRI showing CAVM in 9-month-old patient.
Spinal AVMs are rare, occurring in about 1% of individuals with HHT. They may manifest as subarachnoid hemorrhage, progressive myelopathy, radicular pain, or sphincter disturbance.30 They are often unsuspected, and it may take months or years from onset of symptoms to diagnosis.
Although often clinically silent, hepatic vascular shunts can present as high-output heart failure, portal hypertension, biliary disease, and portosystemic encephalopathy.31–34 Hepatic vascular lesions include intrahepatic shunts of different types and disseminated intraparenchymal telangiectases.32 Hepatic involvement may be more common in women.35,36 It is not yet clear whether hepatic vascular lesions in HHT are congenital and simply present later in life or whether they develop over time. The prevalence of hepatic involvement in HHT is unknown but probably higher than previously recognized. In one study hepatic vascular abnormalities were identified by multi-detector row helical CT in 78% of consecutive HHT patients.33 Most remain asymptomatic.
AVMs have been described rarely in other locations including coronary arteries37,38 and vessels of eye,39,40 spleen,41 urinary tract,42 and vagina.43
MANAGEMENT OF HHT
Many types of specialists care for patients with HHT, thus mul-tidisciplinary HHT specialty centers have evolved nationwide and internationally. Although providing care in the organ system of their expertise, HHT center clinicians are also part of a team knowledgeable about the intricacies of HHT. Ideally, the multi-disciplinary team includes an ENT surgeon, interventional radiologist, pulmonologist, neuroradiologist, neurosurgeon, medical geneticist, genetic counselor, cardiologist and echocardiographer, gastroenterologist, hepatologist, and hematologist. Neurologists and pediatricians also play a role in the team.
Individuals with HHT should receive an initial workup at diagnosis and then preventive care and surveillance. Definitive treatment should be provided if needed. Individuals at risk for HHT should undergo evaluation to determine if they are affected. A list of multidisciplinary specialty clinics for HHT can be found on the web site of the HHT Foundation International (http://www.hht.org).
Surveillance and screening
It is recommended that individuals with HHT have the following:
-
CBC and hematocrit, to be repeated if indicated. Testing will be more frequent in individuals with severe epistaxis and/or GI bleeding.
-
Evaluation for occult blood in stool, especially in middle-aged adults
-
Brain MRI with and without gadolinium to screen for CAVMs once at any age27
-
Contrast echocardiography to screen for pulmonary shunting at least once44–46
-
If shunting is found, chest CT with 3-mm cuts is done to characterize PAVMs.47 PAVMs may grow in size over time, so individuals with smaller PAVMs need to be followed.17 Lifelong periodic surveillance for PAVMs is recommended, but frequency and method depend on age of patient and the clinical situation. AVMs with a feeder vessel of over 3 mm should be treated.48
-
Pulmonary screening recommendations in the first decade are not as well defined. Contrast echocardiography is recommended at about 10 years. Before age 10, a minimum of finger oximetry in supine and sitting positions is recommended every 1 to 2 years. (Because most PAVMs are in the lower lobes, many individuals with PAVMs have higher oxygen saturation when lying than when sitting due to effect of gravity.) Oxygen saturations below 97% should be followed with a contrast echocardiogram.10
-
Patients with evidence of pulmonary shunting by echo-cardiogram are placed on the prophylactic antibiotic regimen of the American Heart Association for prevention of infected emboli.24,49,50
-
Screening for liver involvement is not currently performed on a routine basis in most North America HHT clinics except by listening for bruits.
Treatment
-
Mild epistaxis is best managed conservatively. Humidification and the daily application of nasal lubricants by the patient are often helpful. Few randomized clinical trials have evaluated the various treatments used for nosebleeds in HHT. Careful laser ablation may be the most effective treatment for control of moderate nosebleeds.51,52 Most otolaryngologists experienced with HHT recommend avoiding electric and chemical cautery and transcatheter embolotherapy for treatment of recurrent nosebleeds. Otolaryngologists adept at septal dermoplasty using split thickness skin grafts have reported good results in individuals with severe epistaxis.53
-
Skin lesions usually require no treatment, but if they bleed or the patient wishes it for cosmetic reasons, may be treated by laser.
-
Anemia due to nosebleeds or gastrointestinal bleeding can be controlled with oral or parenteral iron. In some individuals, blood transfusion may be necessary.
-
Gastrointestinal bleeding can be treated medically including iron therapy, ethinyl estradiol/norethindrone, danazol, and aminocaproic acid. If severe, bleeding spots can be localized by endoscopy or angiography, and treated by endoscopic application of a heater probe, bicap, or laser.54
-
PAVMs are safely treated using transcatheter embolization (Fig. 4), by an interventional radiologist who regularly treats HHT patients and is familiar with the risks associated with these high-flow lesions and their treatment.24,49 PAVMs should be embolized before pregnancy, but if not diagnosed early enough, can be treated during the second trimester.55
Fig 4 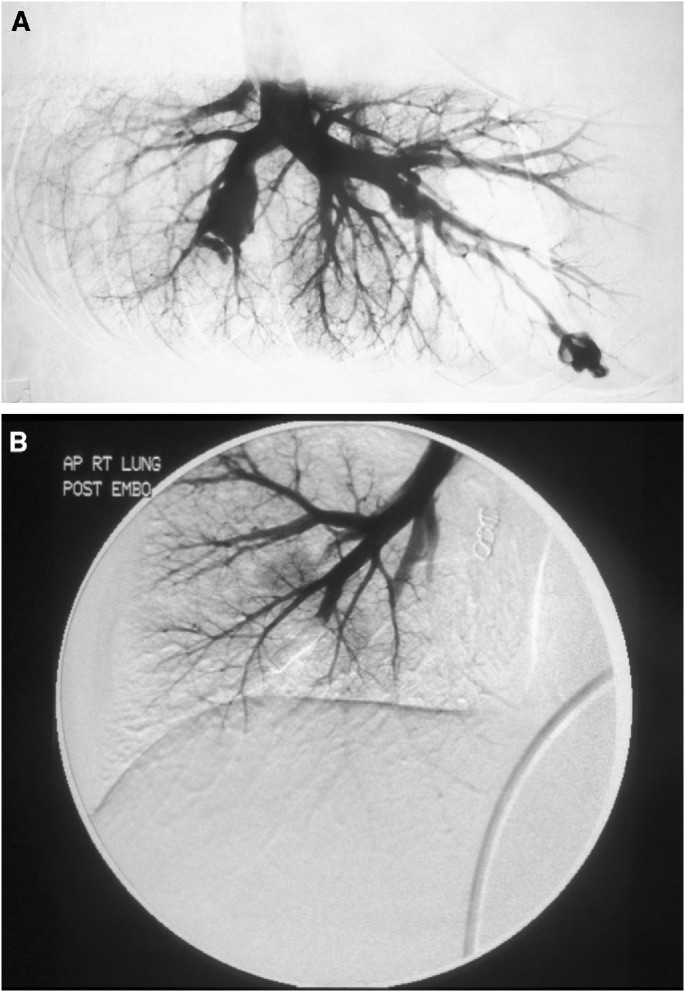
Angiogram showing PAVM preembolization (A) and postembolization (B).
-
Techniques currently used to treat CNS AVMs include transcatheter embolization, resection, and stereotactic radiosurgery, often in combination.56,57 The most effective management of AVMs in children remains controversial; however, the long-term risks of hemorrhage, neurological deficits, or death when treated conservatively are considered unacceptable.7,56
-
Symptomatic liver involvement is difficult to treat, with embolization resulting in a high mortality due to liver infarction.34,58,59 At present, the treatment of choice is liver transplantation.60
-
Medications that interfere with normal coagulation such as aspirin and ibuprofen should be avoided.

PATHOGENESIS OF HHT
HHT is an autosomal-dominant disorder presently linked to two loci. Mutations of the ENG, localized to the long arm of chromosome 9 (9q33-q34.1), cause HHT1 (OMIM: 187300),61–63 whereas HHT2 (OMIM: 600376) is caused by mutations of the gene encoding activin A receptor, type II-like kinase 1 (ACVRL1; also called activin receptor-like kinase 1, ALK1), localized on the long arm of chromosome 12 (12q11-q14).64–66ENG has 14 exons and occupies 30 kb,67 whereas the 10 exons of the ALK1 gene span more than 15 kb of genomic DNA.68
ENG and ALK1 encode receptor proteins, which are members of the transforming growth factor-β (TGFβ) superfamily.69–71 Thus, HHT is caused by a disturbance in the TGFβ signaling pathway. TGFβ signaling is important in the regulation of many cellular processes such as proliferation, differentiation, adhesion, and migration.72 However, the exact mechanism of how a disturbance of this pathway leads to HHT remains unclear.
Various ligands of the TGFβ superfamily bind to TGFβ receptors and form a heteromeric structure. ALK1 encodes a type I TGFβ receptor (TβR1, SwissProt: P37023), which is expressed in endothelial cells (ECs) and highly vascularized tissues such as lung and placenta.69,73,74 Endoglin (also called CD105, SwissProt: P17813) is a homodimeric transmembrane glycoprotein. Its expression is high in ECs, syncytiotropho-blasts, activated monocytes, and tissue macrophages.75,76 Although there is some controversy, endoglin is also known as TβRIII because of its 71% similarity to betaglycan, a TβRIII protein.70,77
TGFβ signaling occurs through the phosphorylation of the Gly/Ser-rich domain (GS domain) of TβRI by TβRII, which then activates downstream signaling via phosphorylation of Smads.69,78–81 Upon activation, Smad4 and its cell type–specific DNA binding partner move into the nucleus, and regulate transcription of the target gene(s).81,82 Interestingly, in a recent article, a few families with a combined syndrome of juvenile polyposis and HHT have been described.83 This condition is caused by mutations in the MADH4 (SMAD4) gene, suggesting the importance of different components of the TGFβ signaling pathway in HHT pathogenesis.
Endoglin is known to bind the heteromeric TβRI and TβRII complex. Because binding of ENG is not necessary for the phosphorylation or the function of the heteromeric complex, it is suggested that ENG modulates the downstream signaling pathway, i.e., Smad signaling.77
A fine balance between ALK1 and ALK5, both of which are TβRI molecules, plays an important role in angiogenesis. Although TGFβ signaling is through ALK5 in many cell types, in ECs, both ALK1 and ALK5 are utilized.84,85 ALK1 is a positive regulator of angiogenesis, whereas ALK5 inhibits proliferation and migration of ECs.85,86 ENG regulates the ALK1/ALK5 balance by favoring ALK1 in ECs.84–86 Hence, mutations in either ENG or ALK1 cause similar findings.
Based on the predominance of mutations leading to premature stop codons and truncation of the ENG protein, a dominant-negative model was initially proposed as the mechanism of disease for HHT.67,77 In this model, the truncated proteins were predicted to interfere with TGFβ signaling either by binding to normal endoglin or by being secreted and sequestering extracellular TGFβ.87 Subsequently, in an expression study of a splice site mutation leading to an inframe deletion of exon 3, ENG expression on the surface of both endothelial cells and monocytes was found to be 50% of controls.88 This result suggests that mutant forms of ENG, when expressed, do not heterodimerize with wild-type or homodimerize and are probably degraded intracelularly.88 The dominant haploinsufficiency (or null allele) mechanism assumed from this study was supported by other independent studies.88–90 In expression studies with six different missense mutations, mutant proteins were not detected at the cell surface, possibly due to misfolding of the protein. However, when these missense mutations were coexpressed with normal ENG, the normal and mutant proteins were able to dimerize and were trafficked to the cell surface.91 Similar studies with two truncation mutations yielded discrepant results, supporting both the haploinsufficiency and the dominant-negative mechanisms.92,93
Using a novel polyclonal antibody, ALK1 protein levels were shown to be reduced in HHT2 patients.94 In addition, some mutations appear to result in undetectable transcript levels, indicating they result in functionally null alleles.68,94,95 The lack of surface expression of the mutant proteins suggests that these mutations lead to structural alterations resulting in misfolding and intracellular degradation of the proteins.95,96,97
In summary, although dominant-negative mechanism cannot be ruled out for some ENG mutations, haploinsufficiency is considered to be the main mechanism leading to HHT.
Animal models of HHT
After the identification of ALK1 and ENG mutations in HHT patients, several research groups established HHT models by gene targeting in mice and zebrafish.
Complete absence of the endoglin gene in the mouse (Eng−/−) is embryonic lethal due to profound cardiovascular defects in the yolk sac vasculature.98 The early steps of vessel formation appear normal in these mice, suggesting that endoglin has a role in angiogenesis rather than vasculogenesis.92,98–100 In heterozygous mice (Eng+/−), development of disease manifestations depends on the genetic background of the mouse, suggesting the role of modifier genes. When present, HHT symptoms show age-dependent manifestations and variable expression. Lack of clinical symptoms in heterozygous mice followed for more than a year suggests a mutant endoglin allele is apparently necessary but not sufficient to cause disease.
Although the targeted disruption of Alk1 in mouse (Alk1−/−) is embryonic lethal, death occurs at a later stage compared to Eng−/−mice. The Alk1−/−mice exhibit defective endothelial remodeling. Heterozygous mice (Alk1+/−) manifest symptoms similar to those seen in HHT patients.101,102
The knock-in Alk1 model showed that the Alk1 protein is preferentially expressed in the endothelium of arteries in early embryogenesis and throughout development.103Alk1 expression was diminished in blood vessels of the adult mice, but was induced in the neovascular endothelium during wound healing, tumor growth, and in the arteries corresponding to increased blood flow.
Recently, zebrafish models of HHT with Eng and Alk1 mutations have been generated that further support that HHT is caused by the haploinsufficiency of either Eng or Alk1 genes.104,105
Human ENG mutations
To date, 114 different mutations in the ENG gene have been reported (Table 1). Only 16 of these mutations were reported in more than one family (14.03%). There are 24 missense mutations (21.05%), 13 nonsense mutations (11.40%), 39 small (< 40 bp) deletions (34.22%), 6 large (>100 bp) deletions, insertions or duplications (5.26%), 15 small insertions (13.16%), 14 splice site mutations (12.28%), and 3 small deletions/insertions (2.63%). The distribution of these mutations in the gene is shown in Fig. 5. These mutations are spread over the first 12 exons and there is no mutation “hot spot.” The g.IVS+1G>A and c.1238G>T mutations, which seem to be frequent, have all been found in the small islands of the Netherlands Antilles with a reported HHT prevalence of at least 1/1300.1 This is thought to be the result of a “founder effect,” with these mutations having been introduced into the African slave population by early Dutch colonists.106

Schematic of genomic structure of ENG (not drawn to scale), indicating the locations of mutations identified to date.
Human ALK1 mutations
Of the 80 different ALK1 mutations reported to date there are 44 missense mutations (55%), 16 deletions (20%), 9 nonsense mutations (11.25%), 7 insertions (8.75%), one duplication (1.25%), one insertion/deletion (1.25%), one insertion/ deletion/ missense mutation (1.25%), and one splice site mutation (1.25%) (Table 2, Fig. 6). Only 20 of these mutations have been reported in more than one family (25%). In general, ALK1 mutations are distributed over the entire coding region, with exons 3, 7, and 8 being most frequently affected (20%, 22.5%, and 25%, respectively;Table 2). Although the c.1111_1112insG mutation was observed in 18 patients not known to be related, they were all from the same region and 17 of them shared the same haplotype.107 Interestingly, no mutations associated with HHT have been reported in exon 5, which encodes the GS domain of ALK1 protein important for phosphorylation. To date, the only mutation in exon 5 (g.536A>C, D179A) was reported in a patient with primary pulmonary hypertension without features of HHT.96

Schematic of genomic structure of ALK1 (not drawn to scale), indicating the locations of mutations identified to date.
The most frequent mutations in the ALK1 gene are missense mutations (55%), whereas frameshift mutations (50%) are commonly observed in the ENG gene. Unlike ENG, large deletions/insertions/duplications have not been reported in the ALK1 gene. Furthermore, splice site mutations have been reported only once in ALK1,107 whereas they account for 12% of ENG mutations.
Mutation frequency
A total of 114 different mutations were observed in ENG, compared to 80 mutations in ALK1. When the possible founder effects are excluded, the percentage of ENG and ALK1 mutations thus far reported as causing HHT is almost the same (53% and 47%, respectively). It should be noted, however, that most affected individuals genotyped to date presented to HHT specialty clinics worldwide. Ascertainment bias related to the morbidity associated with pulmonary involvement cannot be ruled out and would favor the identification of HHT1.
It is difficult to assess the new mutation frequency in HHT. To date, only 4 de novo mutations have been reported in HHT1 patients, with no such reports for HHT2. The age-dependent expression of HHT14,15 might result in underestimation of the mutation frequency. DNA samples analyzed to date have been from either HHT patients or HHT relatives. It is likely that some patients with de novo mutations would be missed due to lack of family history of HHT or not having developed the symptoms to bring them to medical attention.
CLINICAL MOLECULAR ASPECTS OF HHT
Genotype/phenotype correlations
Although some reports indicate that PAVMs108,109 are more frequent in HHT1 and hepatic involvement may be more common in HHT2,35,36,110 HHT is clinically very heterogeneous. Significant intrafamilial as well as interfamilial variations are observed in HHT families. It is not possible to diagnose subtypes of HHT, i.e., HHT1 or HHT2, based on the clinical presentation. All reported manifestations of HHT have been seen in both types. There are individuals in HHT2 families with early and severe presentation of pulmonary AVM, epistaxis, and telangiectases. There are individuals with HHT1 and liver involvement. Therefore, the gene involved cannot be predicted accurately by clinical symptoms.36,111
This has two major implications for molecular diagnosis in HHT and counseling regarding subsequent results. First, the decision to test only the ALK1 or ENG gene in a particular family based on clinical findings will result in the HHT-causing mutation being missed in a substantial number of families. Initial genetic diagnosis in a family should thus usually include analysis of both genes. Secondly, families and individuals with both types of HHT should be counseled regarding their risk for all the vascular lesions and associated symptoms that have been reported in HHT patients.
Neither specific ENG nor specific ALK1 mutations seem to correlate with the severity of the phenotype. The types or the positions of the mutations cannot be used to predict the manifestations or the severity of HHT in an individual.
Molecular diagnostic testing
Molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ALK1 and ENG genes. The lack of common alleles or mutational types in both genes makes the development of simpler and more reliable diagnostic approaches difficult. Sequencing may be targeted following the use of a mutation scanning technology, or may involve sequencing the entire coding region of the endoglin and ALK1 genes.
Mutation detection rate via sequencing is ≈80% to 90%.112 One possible reason for this < 100% rate is the limitation of sequencing in detection of big deletions, insertions, and duplications. Such alterations have been reported in ENG. Cymerman et al.112 reported using quantitative multiplex PCR (QM-PCR) in which an unrelated gene, whose copy number is known, was used as an internal control to determine the copy number of ENG. However, some large insertions, large deletions, or duplications can still remain undetected after use of QM-PCR. Application of other molecular diagnostic techniques such as detecting loss of heterozygosity, long PCRs, quantitative Southern blots, or a combination of several diagnostic techniques might increase the mutation detection rate.
Additionally, mutations in the 5′ and 3′ untranslated regions can account for some of the mutations undetected by sequencing. However, because noncoding regions are not routinely screened by sequencing, mutations in these regions remain undetected.
Locus heterogeneity and existence of another, yet to be identified HHT gene might be another reason for the reduced mutation detection rate. Rare HHT families are apparently not linked to either ALK1 or ENG.11 However, linkage data can be affected by the incorrect assignment of affected versus unaffected status because manifestations of HHT sometimes do not present until middle or later adulthood. Also, symptoms (usually recurrent epistaxis) suggestive of HHT can occur in unaffected individuals in HHT families. In fact, a mutation in ALK1 was described in a family previously reported as nonlinked to HHT2 locus.113 Therefore, the existence of a third locus remains unclear.
Another challenge in genetic diagnosis for HHT is that the detection of a novel missense mutation is very common. Missense mutations, most of them novel, constitute 21% of all mutations identified in the ENG gene and 54% of the mutations identified in the ALK1 gene to date (Tables 1 and 2). It is difficult to determine with certainty that the identified sequence variation is truly disease-causing rather than a benign variant. Family studies, population studies, or expression analyses can be helpful in interpreting these results, but in general, these studies are not practical to perform in a clinical laboratory in the evaluation of a specific case.
Testing protocol and results
Molecular diagnosis of HHT requires a stepwise, family based approach. An individual who meets diagnostic criteria for HHT should be tested first in each family to determine whether the family’s HHT mutation can even be detected. Confirmation of HHT in a relative, who will serve as the index case for the purposes of genetic testing, requires a careful family and medical history by a clinician familiar with HHT. A family history consistent with HHT is one of the most useful clinical diagnostic criteria.
Following American College of Medical Genetics guidelines (ACMG 2000),114 the interpretation of the possible test results from sequencing the coding regions of ENG and ALK1 in an affected relative include the following:
-
1
Sequence variation is previously reported and is a recognized cause of the disorder.
-
2
Sequence variation is previously unreported and is of the type that is expected to cause the disorder.
-
3
Sequence variation is previously unreported and is of the type that may or may not be causative of the disorder.
-
4
Sequence variation is previously unreported and is probably not causative of disease.
-
5
Sequence variation is previously reported and is a recognized neutral variant.
Molecular diagnostic testing for HHT in other relatives is not indicated unless the interpretation of the test result in the family’s index case was either 1 or 2.
Once a causative mutation has been identified in an affected relative, the chance of a conclusive result is much higher and the cost of the analysis much less, given its targeted nature. The cost to test the initial affected relative will typically exceed $1000, whereas the cost to test subsequent relatives for an already identified mutation is typically about several hundred dollars in clinical genetics laboratories.
For a person being tested for a mutation already identified in an affected family member, there are two possible test results: (1) Positive for the family mutation. This person has inherited the DNA sequence variation causing HHT in their family; i.e., this person has HHT. (2) Negative for the family mutation. This person has not inherited the DNA sequence variation causing HHT in their family; i.e., this person does not have HHT.
The three clinical genetics laboratories in North America that currently offer testing for HHT recommend that molecular diagnostic testing be coordinated and ordered through one of the HHT Specialty Centers, listed on the HHT Foundation International web site (http://www.hht.org), or a genetics professional. This recommendation is due to the family-based, multiple-step approach required for testing and the complexity of the results and their significance. Primary care physicians often lack the expertise to appropriately order and interpret sequencing-based genetic tests.115 Involvement of a specialist also helps ensure optimal medical management based on the genetic test results.
Genetic counseling considerations
The medical and genetic aspects of HHT and its molecular diagnosis are complex. However, compared with many genetic disorders, the ethical issues surrounding genetic testing for HHT are less complex. The disorder is very difficult to diagnose based on clinical examination and medical history in the first few decades of life, and there is no “nongenetic” diagnostic laboratory test. Yet there are treatable congenital internal lesions that cannot be found on external examination that can present suddenly and catastrophically.
In general, the benefits of genetic testing are assumed to outweigh the risks if current medical management would be changed as a result of testing. Based on the criterion of early, preventable risks, it is reasonable to offer molecular genetic diagnosis for HHT early in life for individuals who are at 50% risk. For example, in a family with an identified HHT mutation, sending umbilical cord blood for mutation analysis on an at-risk newborn infant is indicated.
Usual concerns related to presymptomatic genetic testing and genetic testing in childhood are not significant issues in HHT molecular diagnosis. It can be argued that “presymptomatic testing” as it is usually thought of, does not exist in this disorder. The absence of manifestations or symptoms detectable by clinical examination and history in a 1-month-old whose parent has HHT offers almost no reassurance that he/ she is unaffected and without risk from a CAVM.
An individual who is to serve as the “known affected,” index case for molecular diagnosis in a family should understand that this analysis of their sample will not significantly alter their course or care, but will most likely confirm their clinical diagnosis. A major purpose of testing him/her is to determine whether genetic diagnosis will be possible in other at-risk relatives. The purpose of the testing, and thus the content of pre-test genetic counseling, is very different for the index case than for the other relatives who are subsequently tested. The index cases should also understand the range of possible test results from sequencing-based genetic testing. These include a possible “negative” result despite a clinical diagnosis of HHT and identification of a sequence variation of uncertain significance. In either case, the result of their testing would not allow for the desired accurate molecular genetic diagnostics for at-risk relatives.
Potential risks of genetic testing are emotional and practical in nature. Risks of an emotional nature include negative feelings about being labeled as having a genetic disorder, fear of the future, or in the case of a parent, guilt about having passed a hereditary condition to a child. Possible practical risks include noninsurability once the diagnosis of a genetic disorder has been confirmed. Many states have enacted genetic nondis-crimination legislation, but the laws are quite varied in their focus and scope. Professional genetics organizations support federal legislation to assure individuals and families that neither health care coverage nor employment status will be jeopardized by genetic test results.
SUMMARY
HHT is inherited as an autosomal-dominant condition with high penetrance and extremely variable age-dependent expression. Locus heterogeneity exists with 2 molecular forms described to date. Although there are likely overall differences between HHT1 and HHT2 in frequency and severity of certain manifestations, they are not distinct enough to alter clinical management based on the specific genetic form of HHT.
Molecular genetic diagnosis for HHT is complex. There is a substantial chance that genetic testing, even in a clearly affected individual, will result in a “negative” result or “sequence variation of uncertain significance.”
Despite its complexities and limitations, molecular diagnostic testing for HHT is available and indicated for individuals with symptoms strongly suggestive of HHT, as well as asymptomatic individuals with an affected first-degree relative. Affected individuals of any age, even those currently asymptomatic for HHT based on clinical examination and medical history, may benefit from having their diagnosis confirmed by molecular genetic testing. Testing in any particular family should begin with a clinically confirmed affected individual, not one in whom the diagnosis is in question. This requires a multigenerational, family-based approach to HHT molecular diagnosis.116–123
References
Westermann CJ, Rosina AF, De Vries V, de Coteau PA . The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: a family screening. Am J Med Genet 2003; 116A: 324–328.
Kjeldsen A, Vase P, Green A . Hereditary haemorrhagic telangiectasia: a population-base study of prevalence and mortality in Danish patients. J Int Med 1999; 245: 31–39.
Plauchu H, Bideau A . Epidemiologie et constitution dun registre de population a propos dune concentration geographique dune maladie hereditaire rare. Population 1984; 39: 765–786.
Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, et al. Genetics epidemiology of hereditary hemorrhagic telangiectasia in a local community in the Northern Part of Japan. Hum Mutat 2002; 19: 140–148.
Morgan T, McDonald J, Anderson C, Ismail M, Miller F, Mao R, et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia. Pediatrics 2002; 109: 1–7.
Roman G, Fisher M, Perl D, Poser C . Neurological manifestations of hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber Disease): Report of 2 cases and review of the literature. Ann Neurol 1978; 4: 130–144.
Kondziolka D, Humphreys R, Hoffman H, Hendrick EB, Drake J . Arteriovenous malformations of the brain in children: a forty year experience. Can J Neurol Sci 1992; 19: 40–45.
Mitchell R, Austin E . Pulmonary arteriovenous malformation in the neonate. J Pediatr Surg 1993; 28: 1536–1538.
Reichman H, Sorensen B . Excision of spinal cord arteriovenous malformation. Rocky Mt Med J 1971; 68: 21–24.
Guttmacher A, Marchuk D, White R . Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333: 918–924.
Wallace G, Shovlin C . A hereditary haemorrhagic telangiectasia family with pulmonary involvement is unlined to the known HHT genes, endoglin and ALK-1. Thorax 2000; 55: 685–690.
Shovlin C, Guttmacher A, Buscarini E, Faughnan M, Hyland R, Westermann C, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber Syndrome). Am J Med Genet 2000; 91: 66–67.
Assar A, Friedman C, White R . The natural history of epistaxis in hereditary hemorrhagic telangiectases. Laryngoscope 1991; 101: 977–980.
Plauchu H, de Chadarevian JP, Bideau A, Robert JM . Age-related clinical profile of hereditary haemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32: 291–297.
Porteous M, Burn J, Proctor S . Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet 1992; 29: 527–530.
Kjeldsen A, Kjeldsen J . Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol 2000; 95: 415–418.
Vase P, Holm M, Arendrup H . Pulmonary arteriovenous fistulas in hereditary hemorrhagic telangiectasia. Acta Med Scand 1985; 218: 105–109.
Haitjema T, Disch F, Overtoom T, Westermann C, Lammers J . Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med 1995; 99: 519–524.
Kjeldsen A, Oxhoj H, Andersen P, Green A, Vase P . Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (HHT). J Int Med 2000; 248: 255–262.
Shovlin C, LeTarte M . Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54: 714–729.
White R . Pulmonary arteriovenous malformations: how do we diagnoses them and why is it important to do. soRadiology 1992; 182: 633–635.
Ference B, Shannon T, White R, Zawin M, Burdge C . Life-threatening pulmonary hemorrhage with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Chest 1994; 106: 1387–1390.
Moussouttas M, Fayad P, Rosenblatt M, Hashimoto M, Pollak J, Henderson K, et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology 2000; 55: 959–964.
White R, Pollak J, Wirth J . Pulmonary arteriovenous malformations: diagnosis and transcatheter embolotherapy. J Vasc Interv Radiol 1996; 7: 787–804.
Hewes R, Auster M, White R . Cerebral embolism: first manifestation of pulmonary arteriovenous malformation in patients with hereditary hemorrhagic telangiectasia. Cardiovasc Inter Rad 1985; 8: 151–155.
Shovlin C, Winstock A, Peters A, Jackson J, Hughes J . Medical complications of pregnancy in hereditary hemorrhagic telangiectasia. Q J Med 1995; 88: 879–887.
Fulbright R, Chaloupka J, Putman C, Sze G, Merriam M, Lee G, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neurol Radiol 1998; 19: 477–484.
Matsubara S, Manzia J, ter Brugge K, Willinsky R, Montanera W, Faughnan M . Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. Am J Neuroradiology 2000; 21: 1016–1020.
Willemse R, Mager J, Westermann C, Overtoom T, Mauser H, Wolbers J . Bleeding risk of cerebrovascular malformation in hereditary hemorrhagic telangiectasia. J Neurosurg 2000; 92: 779–784.
MontAlverne F, Musacchio M, Tolentino V, Belzile F, Riquelme C, Tournade A . Giant spinal perimedullary fistula in hereditary haemorrhagic telangiectasia: diagnosis, endovascular treatment and review of the literature. Neuroradiology 2003; 45: 830–836.
Garcia-Tsao G, Korzenik J, Young L, Henderson K, Jain D, Byrd B, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343: 931–936.
Memeo M, Stabile Ianora AA, Scardapane A Buonamico P, Sabba C, Angelelli G . Hepatic involvement in hereditary hemorrhagic telangiectasia: Abdom Imaging 2004. Epub ahead of print.
Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G . Hereditary hemorrhagic telangiectasia: multidetector row helical T assessment. Radiology 2004; 230250–230259.
Saluja S, White RI . Hereditary hemorrhagic telangiectasia of the liver; Hyperper-fusion with relative ischemia - poverty amidst plenty?. Radiology 2004; 230: 25–27.
Buscarini E, Buscarini L, Danesino C, Pinatanida M, Civardi G, Quaretti P, et al. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia: Doppler sonographic screening in a large family. J Hepato 1997; 26: 111–118.
McDonald J, Miller F, Hallam S, Nelson L, Marchuk D, Ward K . Clinical manifestations in a large hereditary hemorrhagic telangiectasia type 2 kindred. Am J Med Genet 2000; 93: 320–327.
His DH, Ryan GF, Hellems SO Cheeran DC, Sheils LA . Large aneurysms of the ascending aorta and major coronary arteries in a patient with hereditary hemorrhagic telangiectasia. Mayo Clin Proc 2003; 78: 774–776.
Tsuiki K, Tamada Y, Yasui S . Coronary artery aneurysm without stenosis in association with Osler-Weber-Rendu disease: A case report. Angiology 1991; 42: 55–58.
Brant A, Schachat A, White R . Ocular manifestations in hereditary hemorrhagic telangiectasia. Am J Ophthalmol 1989; 107: 642–646.
Vase I, Vase P . Ocular lesions in hereditary hemorrhagic telangiectasia. Acta Ophthalmol 1979; 57: 1084–1090.
Lande A, Bedford A, Schechter LS . The spectrum of arteriographic findings in Osler-Weber- Rendu disease. Angiology 1976; 27: 223–240.
Ziani M, Valignat C, Lopez J, Ruffion A, Plauchu H, Perrin P . Renal arteriovenous malformation requiring surgery in Rendu-Osler-Weber Disease. J Urololgy 2000; 164: 1292–1293.
Humphries JE, Frierson HF, Underwood PB . Vaginal telangiectasias: unusual presentation of the Osler-Weber-Rendu syndrome. Obstet Gynecol 1993; 81: 865–866.
Barzilai B, Waggoner A, Spessert C, Picus D, Goodenberger D . Two-dimensional contrast echocardiography in the detection and follow-up of congenital pulmonary arteriovenous malformations. Am J Cardiol 1991; 68: 1507–1510.
Kjeldsen A, Oxhoj H, Andersen P, Elle B, Jacobsen J, Vase P . Screening Procedures and pulmonary angiography in patients with hereditary hemorrhagic telangiectasia. CHEST 1999; 116: 432–439.
Nanthakumar K, Graham A, Robinson T, Grande P, Pugash R, Clarke J, et al. Contrast echocardiography for detection of pulmonary arteriovenous malformations. Am Heart J 2001; 141: 243–246.
Remy J, Remy-Jardin M, Wattinne L, Deffontaines C . Pulmonary arteriovenous malformations: Evaluation with CT of the chest before and after treatment. Radiology 1992; 182: 809–816.
White R, Pollak J . Pulmonary arteriovenous malformations: diagnosis with three-dimensional helical CT: A breakthrough without contrast media. Radiology 1994; 191: 613–614.
Lee D, White R, Egglin T, Pollak J, Fayad P, Wirth J, et al. Embolotherapy of large pulmonary arteriovenous malformations: long-term results. Ann Thorac Surg 1997; 64: 930–940.
Christenson G . Nosebleeds may mean something much more serious: an introduction to HHT. JADA 1998; 129: 635–637.
Parkin J, Dixon JA . Laser photocoagulation in hereditary hemorrhagic telangiectasia. Otolaryngol Head Neck Surg 1981; 89: 204–208.
Shapshay S, Oliver P . Treatment of hereditary hemorrhagic telangiectasia by Nd-Yag laser photocoagulation. Laryngoscope 1984; 94: 1554–1556.
Saunders W . Septal dermoplasty for control of nosebleeds caused by hereditary hemorrhagic telangiectasia or septal perforations. Trans Am Acad Ophthalmol Otolaryngol 1960; 64: 500–506.
Longacre A, Gross C, Gallitelli M, Henderson K, White R, Proctor D . Diagnosis and management of gastrointestinal bleeding in patient with hereditary hemorrhagic telangiectasia. Am J Gastroenterology 2003; 98: 59–65.
Gershon A, Faughnan M, Chon K, Pugash R, Clark J, Bohan M, et al. Transcatheter embolotherapy of maternal pulmonary arteriovenous malformations during pregnancy. CHEST 2001; 119: 470–477.
Levy EI, Niranjan M, Thompson T, Scarrow A, Kondziolka D, Flickinger J, et al. Radiosurgery for childhood intracranial arteriovenous malformations. Neurosurgery 2000; 47: 834–842.
Vinuela F, Dion J, Duckwiler G, Martin N, Lylyk P, Fox A, et al. Combined endovascular embolization and surgery in the management of cerebral arteriovenous malformations: experience with 101 cases. J Neurosurg 1991; 75: 856–864.
Odorico J, Hakim M, Becker Y . Liver transplantation as definitive therapy for complication after arterial embolization for hepatic manifestations of hereditary hemorrhagic telangiectasia. Liver Transpl Surg 1998; 4: 483–490.
Miller F, Whiting J, Korzenik J, White R . Caution with use of hepatic embolization in the treatment of hereditary hemorrhagic telangiectasia. Radiology 1999; 213: 928–930.
Boillot O, Bianco F . Viale J, Liver transplantation resolves the hyperdynamic circulation in hereditary hemorrhagic telangiectasia with hepatic involvement. Gastroenterology 1999; 116: 187–192.
Fernandez-Ruiz E, St-Jacques S, Bellon T, Letarte M, Bernabeu C . Assignment of the human endoglin gene (END) to 9q34 qter. Cytogenet Cell Genet 1993; 64: 204–207.
McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, et al. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat Genet 1994; 6: 197–204.
Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet 1994; 6: 205–209.
Vincent P, Plauchu H, Hazan J, Faure S, Weissenbach J, Godet J . A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Gen 1995; 4: 945–949.
Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA, et al. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res 1995; 5: 21–28.
Roijer E, Miyazono K, Astrom AK, Geurts van Kessel A, ten Dijke P, Stenman G . Chromosomal localization of three human genes encoding members of the TGF-beta superfamily of type I serine/threonine kinase receptors. Mamm Genome 1998; 9: 266–268.
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994; 8: 345–351.
Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, et al. The Activin receptor-like kinase 1 gene: Genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61: 60–67.
Attisano L, Carcamo J, Venture F, Weis FM, Massague J, Wrana JL . Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993; 5: 671–680.
Cheifetz S, BellÓn T, Calés C, Vera S, Bernabeu C, Massague J, et al. Endoglin is a component of the transforming growth factor- receptor system in human endothelial cells. J Biol Chem 1992; 267: 19027–19030.
Bellon T, Corbi A, Lastres P, Cales C, Cebrian M, Vera S, et al. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur J Immunol 1993; 23: 2340–2345.
Attisano L, Wrana JL . Signal transduction by the TGF- superfamily. Science 2002; 296: 646–1647.
ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, et al. Activin receptor-like kinases: a novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene 1993; 8: 2879–2887.
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13: 189–195.
Gougos A, St Jacques S, Greaves A, O’Connell PJ, d’Apice AJ, Buhring HJ et al. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int Immunol 1992; 4: 83–92.
Lastres P, Bellon T, Cabanas C, Sanchez-Madrid F, Acevedo A, Gougos A, et al. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur J Immunol 1992; 22: 393–397.
Barbara NP, Wrana JL, Letarte M . Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor- superfamily. J Biol Chem 1999; 274: 584–594.
Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992; 71: 1003–1014.
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J . Mechanism of activation of the TGF-beta receptor. Nature 1994; 370: 341–347.
Wieser R, Wrana JL, Massague J . GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J 1995; 14: 2199–2208.
Massagué J . TGF- signal transduction. Annu Rev Biochem 1998; 67: 753–791.
Derynck R, Zhang Y, Feng XH . Smads: Transcriptional activators of TGF- responses. Cell 1998; 95: 737–740.
Gallione CJ, Repetto GM, Legius E, Rustig AK, Schelley SL, Tejpar S, et al. A combined syndrome of juvenile polyposis with mutations in MADH4 (SMAD4). Lancet 2004; 363: 852–859.
Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A 2000; 97: 2626–2631.
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P . Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 2002; 21: 1743–1753.
Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ ALK5 signaling. Mol Cell 2003; 12: 817–828.
McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteus AE, et al. Six novel mutations in the endoglin gene in hereditary hemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum Mol Genet 1995; 4: 1983–1985.
Pece N, Vera S, Cymerman U, White R, Wrana J, Letarte M . Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest 1997; 100: 2568–2579.
Pece-barbara N, Cymerman U, Vera S, Marchuk D, Letarte M . Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet 1999; 8: 2171–2181.
Paquet ME, Pece-Barbara N, Vera S, Cymerman U, Karabegovic A, Shovlin C, et al. Analysis of several endoglin mutants reveals no endoglins mature or secreted protein capable of interfering with normal endoglin function. Hum Mol Genet 2001; 10: 1347–1357.
Lux A, Gallione CJ, Marchuk DA . Expression analysis of endoglin missense and truncation mutations: insights into protein structure and disease mechanisms. Hum Mol Genet 2000; 9: 745–755.
Bourdeau A, Cymerman U, Paquet ME, Meschino W, McKinnon WC, Guttmacher AE, et al. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformation of patients with hereditary hemorrhagic telangiectasia type 1. Am J Path 2000; 156: 911–923.
Shovlin CL, Hughes JM, Scott J, Seidman CE, Seidman JG . Characterization of endoglin and dentification of novel mutations in hereditary hemorrhagic telangi-ectasia. Am J Hum Genet 1997; 61: 68–79.
Abdalla SA, Pece-Barbara N, Vera S, Tapia E, Paez E, Bernabeu C, et al. Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum Mol Genet 2000; 9: 1227–1237.
Klaus DJ, Gallione CJ, Anthony K, Yeh EY, Yu J, Lux A, et al. Novel missense and frameshift mutations in the activin receptor-like kinase-1 gene in hereditary hemorrhagic telangiectasia. Mutations in brief no. 164. Online. Hum Mutat 1998; 12: 137
Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003; 40: 865–871.
Abdalla SA, Cymerman U, Johnson RM, Deber CM, Letarte M . Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur J Hum Genet 2003; 11: 279–287.
Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, et al. Defective angiogenesis in mice lacking endoglin. Science 1999; 284: 1534–1537.
Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, et al. Endoglin, an ancillary TGF beta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 2000; 217: 42–53.
Torsney E, Charlton R, Diamond AG, Burn J, Soames JV, Arthur HM . Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation 2003; 107: 1653–1657.
Urness LD, Sorensen LK, Li DY . Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 2000; 26: 328–331.
Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, et al. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet 2003; 12: 473–482.
Seki T, Yun J, Oh P . Arterial endothelium-specific activin receptor-like kinase1 expression suggests its role in arterialization and vascular remodeling. Circu Res 2003; 3: 1–9.
Bourdeau A, Dumont DJ, Letarte M . A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104: 1343–1351.
Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 2002; 129: 3009–3019.
Gallione C, Scheessele EA, Reinhardt D, Duits AJ, Berg JN, Westermann CJ, et al. Two common endoglin mutations in families with hereditary hemorrhagic telangiectasia in the Netherlands Antilles: evidence foe a founder effect. Hum Genet 2000; 107: 40–44.
Lesca G, Plauchu H, Coulet F, Lefebvre S, Plessis G, Odent S, et al. Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Hum Mutat 2004; 23: 289–299.
Berg J, Guttmacher A, Marchuk D, Porteous M . Clinical heterogeneity in hereditary hemorrhagic telangiectasia: are pulmonary arteriovenous malformation more common in families linked to endoglinJ Med. Genet 1996; 33: 256–257.
Heutink P, Haitjema T, Breedveld G, Janssen B, Sandkuijl L, Bontekoe C, et al. Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J Med Genet 1994; 31: 933–936.
Olivieri C, Mira E, Delu G, Pagella F, Zambelli A, Malvezzi L, et al. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet 2002; 39: E39.
Abdalla S, Geisthoff U, Bonneau D, Plauchu H, McDonald J, Kennedy S, et al. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J Med Genet 2003; 40: 494–502.
Cymerman U, Vera S, Amna K, Abdalla S, Letarte M . Characterization of 17 novel endoglin mutations associated with hereditary hemorrhagic telangiectasia. Hum Mutat 2003; 21: 482–492.
Piantanida M, Buscarini E, Dellavecchia C, Minelli A, Rossi A, Buscarini L, et al. Hereditary haemorrhagic telangiectasia with extensive liver involvement is not caused by either HHT1 or HHT2. J Med Genet 1996; 33: 441–443.
American College of Medical Genetics recommendations for standards for interpretation of sequence variations Genet. Med 2000; 2: 302–303.
Menasha J, Schechter C, Willner J . Genetic testing: A physicians perspective. Mt Sinai J Med 2000; 67: 144–155.
Gallione C, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB, et al. Mutation and expression analysis of the endoglin gene in hereditary hemorrhagic telangiectasia reveals null alleles. Hum Mutat 1998; 11: 286–294.
Cymerman U, Vera S, Pece-Barbara N, Bourdeau A, White R, Dunn Jr J, et al. Identification of hereditary hemorrhagic telangiectasia type 1 in newborns by pro-twin expression and mutation analysis of endoglin. Pediat Res 2000; 47: 24–35.
Lastella P, Sabba C, Lenato GM, Resta N, Lattanzi W, Gallitelli M, et al. Endoglin gene mutations and polymorphisms in Italian patients with hereditary haemorrhagic telangiectasia. Clin Genet 2003; 63: 536–540.
Yamaguchi H, Azuma H, Shigekiyo T, Inoue H, Saito S . A novel missense mutation in the endoglin gene in hereditary hemorrhagic telangiectasia. Thromb Haemost 1997; 77: 243–247.
Hereditary Hemorrhagic telangiectasia mutation database, Edinburg, Molecular Genetics Service 2003 Available at: http://137.195.14.43/genisysDR/NVC/198/Display/index.htm.
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345: 325–334.
Kjeldsen AD, Brusgaard K, Poulsen L, Kruse T, Rasmussen K, Green A, et al. Mutations in the ALK-1 gene and the phenotype of hereditary hemorrhagic telangiec-tasia in two large Danish families. Am J Med Genet 2001; 98: 298–302.
D'Abronzo FH, Swearingen B, Klibanski A, Alexander JM . Mutational analysis of activin/transforming growth factor-beta type I and type II receptor kinases in human pituitary tumors. J Clin Endocrinol Metab 1999; 84: 1716–1721.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Bayrak-Toydemir, P., Mao, R., Lewin, S. et al. Hereditary hemorrhagic telangiectasia: An overview of diagnosis and management in the molecular era for clinicians. Genet Med 6, 175–191 (2004). https://doi.org/10.1097/01.GIM.0000132689.25644.7C
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000132689.25644.7C
Keywords
This article is cited by
-
Neuraxial Techniques in Obstetric Patients with Intracranial Lesions
Current Obstetrics and Gynecology Reports (2023)
-
Infective endocarditis with Osler’s nodule in a patient with Osler’s disease: a case report and review of the literature
Journal of Medical Case Reports (2022)
-
Anesthetic considerations for cesarean delivery in a parturient with hereditary hemorrhagic telangiectasia: a case report
JA Clinical Reports (2021)
-
Genetic and epigenetic mechanisms in the development of arteriovenous malformations in the brain
Clinical Epigenetics (2016)
-
A long diagnostic delay in patients with Hereditary Haemorrhagic Telangiectasia: a questionnaire-based retrospective study
Orphanet Journal of Rare Diseases (2012)
