
Abstract
Purpose: Derive estimates of analytic sensitivity and specificity of DNA testing for cystic fibrosis in the United States.
Methods: Analyze published results of the American College of Medical Genetics (ACMG)/College of American Pathologists (CAP) Molecular Genetics Survey between 1996 and 2001, taking into account difficult, simulated clinical samples included for educational purposes.
Results: Analytic sensitivity is 97.9% [95% confidence interval (CI) 96.8–98.7%], and analytic specificity is 99.4% (95% CI 98.7–99.9%) after removing challenges involving delI507 and performing other adjustments. Analytic sensitivity is consistent over the 6 years. Specificity was lower in 1997.
Conclusion: These preliminary estimates indicate that analytic validity of cystic fibrosis testing is very good and can likely be improved. To date, fewer than half of the mutations in the panel recommended for preconceptional or prenatal screening have been challenged. The present study highlights the value of performing confirmatory testing when a mutation is identified to reduce false-positive results.
Similar content being viewed by others


Main
In 2000, the Secretary’s Advisory Committee on Genetic Testing (SACGT) recommended that any DNA or related laboratory test used in routine medical practice be formally evaluated to assess its analytic and clinical validity.1 There is a special urgency to implementing this recommendation when the test is in widespread use in the general population for screening purposes. Such is the case with prenatal screening for cystic fibrosis via carrier testing, which is now becoming widely available following the issuance of guidelines by the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics (ACMG).2,3 The present analysis focuses on the analytic validity of this prenatal screening test, which measures the ability of the laboratory test to accurately identify specific cystic fibrosis mutations.
Analytic validity can be summarized by the sensitivity and specificity of the laboratory methodology, keeping in mind that the effects of pre- and postanalytic steps are included in these summary statistics. Analytic sensitivity is defined as the proportion of positive test results correctly reported from the laboratory among samples containing a mutation that the laboratory’s test is designed to detect. Mutations not detected under these circumstances are labeled “false negatives.” False-negative results can occur during the analytic phase (e.g., sample mix-up, reaction failure due to expired reagents) or in the pre- or postanalytic phases (e.g., sample mix-up, mislabeling, data entry error, inaccurate reading or recording of results, inaccurate interpretation). Analytic specificity is defined as the proportion of negative test results correctly reported by the laboratory when no detectable mutation is present. As with false-negative results, false-positive results can arise in either the analytic phase (e.g., contamination, nonspecific reactions) or in the pre- or postanalytic phases. A third type of error occurs when a mutation is correctly recognized as being present but is incorrectly identified. In the following analyses, wrong mutations are considered false-positive results because there is an opportunity for correcting them by confirmatory testing.
Few data sources exist for reliably estimating analytic validity. Published reports of method comparisons and screening experiences provide limited information on only a few testing methodologies from only a small number of laboratories. In addition, the “true” genotypes of the tested samples are often uncertain because they have not been confirmed by another methodology, laboratory consensus, or direct sequencing. The external proficiency testing program carried out by the ACMG and the College of American Pathologists (CAP) provides a source of data for the present analysis that has several advantages.4 Nearly all clinical testing laboratories in the United States participate. They represent the range of methodologies presently being used. In addition, the sample challenges have confirmed genotypes. However, basing analytic performance estimates on the ACMG/CAP program data also has drawbacks. These include the over-representation of “difficult” samples due to the educational nature of the program, mixing of screening and diagnostic exercises, the “artificial” nature of sample preparation, shipping and handling, and the inclusion of laboratories from outside the United States, as well as reagent manufacturers or research laboratories that do not provide clinical services. One additional consideration might be that laboratories perform differently when testing proficiency samples than when routinely testing clinical samples even though CLIA regulations require proficiency samples to be tested in the same manner as patient samples. This difference might take the form of less good performance because it is not possible to handle the sample according to the routine laboratory protocol (the original sample is extracted DNA rather than blood or buccal scrapings). Alternatively, the performance might be better because the sample might be recognized by laboratory personnel to be for the purpose of evaluating laboratory performance. Despite these shortcomings, data from the ACMG/CAP external proficiency testing program can be useful in establishing an estimate of baseline performance for laboratories testing for cystic fibrosis mutations.
MATERIALS AND METHODS
As part of ACMG/CAP external proficiency testing for cystic fibrosis, purified DNA from established cell lines is distributed to enrolled laboratories. Biyearly reports from the Molecular Genetics Resource Committee are the source of all data used in the analyses.4 Raw data were collected from published tables, and the associated written comments nearly always allowed differentiation between laboratory reporting error and laboratories not testing for the specific mutation challenged. For example, in the 1998-A survey, sample MGL-05 had a genotype of delF508/621 + 1G>T. Eight laboratories reported the genotype as delF508/none of tested, but only three of them routinely tested for the 621 + 1G>T mutation. Only these three laboratories were considered to have reported false-negative results. Analysis was performed by treating results from each allele separately. For example, in the MGL-05 sample, a laboratory that only tested for delF508 would be counted as having one allele challenged for sensitivity (the delF508) and one allele challenged for specificity (the 621 + 1G>T is treated as wild type in this instance). Ninety-five percent confidence intervals (CI) were computed using the binomial distribution (True EPISTAT, Richardson, TX).
RESULTS
Error rates for laboratories participating in the ACMG/CAP external proficiency testing scheme
Table 1 shows the number of alleles tested and the results from the ACMG/CAP Molecular Genetics Laboratory (MGL) Survey from 1996 to 2001. Overall, 97.0% (2131 of 2198 alleles) were correctly identified (95% CI 96.1–97.6%). A complete listing of the sample challenges, the types of errors, and adjustments made during the analysis is available via the ArticlePlus feature at the Genetics in Medicine Web site, http://www.geneticsinmedicine.org. More errors (56) occurred between 1996 and 1998 than between 1999 and 2001 (11). However, the composition of challenges in the earlier time period (i.e., a higher proportion of samples with mutations) explains much of this excess and is taken into account in analyses that are presented later. These error rates are similar to those reported by a similar external proficiency testing program in Europe.5–7
Analytic sensitivity
Because an important aim of external proficiency testing is education and laboratory improvement, reliable analytic performance estimates require that this aspect of these exercises be taken into account. For example, 12% of the challenges (3/25) tested participating laboratories for their ability to distinguish between the delI507 and delF508 mutations. Two of these three challenges occurred in the first 2 years of the survey. The delI507 mutation is expected to occur in less than 1 in 2500 non-Hispanic Caucasians.8 Despite this, it was repeatedly challenged because of the known technical difficulty in making a correct identification. Although errors associated with delI507 would be expected to occasionally occur in practice, these challenges are removed from the final calculations in the present analysis to improve the applicability of the findings in the context of routine testing. An additional complicating feature arises because some “false negatives” are due to laboratories not testing for the mutation. The present analysis takes this into account by classifying the result as a false-negative only if the laboratory is known to test for that mutation.
Table 2 shows the revised analytic sensitivity estimates for individual years and for the overall 6-year time period. Although the number of participating laboratories has remained relatively constant (Table 1), the number of mutational challenges varies widely from a high of 285 in 1998 to a low of 43 in 2000. These differences are due to the genotypes of the samples distributed. Among the seven samples from the first 3 years, all contained at least one mutation. Two were heterozygotes, four were compound heterozygotes, and one was homozygous. Among the 15 samples from the second 3 years, three were heterozygotes, two were compound heterozygotes, and one was homozygous. The remaining eight challenges included no detectable mutations. The yearly estimates of analytic sensitivity vary from a low of 95.3% in 2000 to a high of 100% in 1999. Overall, the rates do not improve significantly over the 6-year time period (χ2 test for trend = 2.2, P = 0.14). The overall consensus of 97.9% is, therefore, a reasonable estimate of analytic sensitivity.
Analytic specificity
Table 3 shows the analytic specificity estimates by individual years and for the overall 6-year time period. The majority of information was collected in the last 3 years. The yearly estimates of analytic specificity vary from a low of 82.5% in 1997 to a high of 100% in 1998 and 2000. There is a highly significant trend toward improved performance (χ2 test for trend = 27, P < 0.001). This effect is due mainly to the high rate of errors in 1997. Sample mix-up among the three challenges is not a plausible explanation for these errors because several of the wrong mutations and false-positive results were mutations that were not present in the samples being tested.
The overall estimate needs to take into account the relative rarity of a wrong mutation in routine screening samples compared with proficiency testing samples. The opportunity for a laboratory to identify a wrong mutation is considerably greater in proficiency testing exercises than in practice. For that reason, the rate of wrong mutations in proficiency testing needs to be adjusted downward. About 1 in 2 chromosomes in the proficiency testing samples have detectable mutations, but only about 1 in 60 chromosomes in non-Hispanic Caucasians will have detectable mutations. Wrong mutations are, therefore, about 30 times more likely to occur as part of proficiency testing than in screening practice. Thus, although Table 3 shows a ratio of 4 false-positive results to 11 wrong mutations, the expected ratio in the general population would be more like 4 false-positive results to less than 1 wrong mutation (11/30). After the rate of “wrong mutation” in the general population is taken into account, the revised estimate of analytic specificity is 99.4% (95% CI 98.7–99.8%).
DISCUSSION
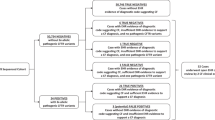
Figure 1 places the analytic sensitivity of 97.9% in perspective by assuming that sequential screening is performed on 10,000 non-Hispanic Caucasian women with a carrier rate of 1 in 25 (4% or 0.04). In this hypothetical cohort, there would be 400 carrier women, and 352 (88%) would be detectable using the recommended 25 mutation panel.9 Only 344 of the 352 detectable carrier women (97.9%), however, would be correctly classified. Confirmatory testing is ordinarily performed only when a mutation is found. Therefore, the eight false-negative results (2.1% of all detectable carrier women) would not be identified, and these errors would not be corrected. If the carrier rate were lowered to 1 in 30, and only 80% of mutations were detectable, there would be six (rather than eight) false-negative results per 10,000 non-Hispanic Caucasian women tested.

Flow diagram describing the analytic sensitivity of cystic fibrosis mutation testing in a population of 10,000 non-Hispanic Caucasian women. The analysis assumes a carrier frequency of 1 in 25. It also assumes that 88% of the mutations are detectable by the testing panel and that 97.9% of the detectable mutations will be correctly identified (analytic sensitivity).
Although the analytic false-negative rate of 2.1% may seem high, it would only rarely lead to a clinical situation that would reveal the error. The most likely situation would be the genotyping of an affected child after the parents had undergone prenatal screening and were identified as not being a carrier couple. If the child did have two mutations for which the laboratory tested, the major cause (besides nonpaternity) would be a false-negative test result in one of the partners. One such case was reported among the 25,000 couples screened in Edinburgh, Scotland.10 The child was diagnosed with cystic fibrosis at 23 months and was found to be a delF508 homozygote. The mother’s test result was a false-negative and, therefore, the partner’s sample was not tested. On the basis of an analytic false-negative rate of 2.1%, this scenario would be expected about once in every 154,000 couples tested (the woman is a carrier of a detectable mutation, but is falsely negative 0.04 * 0.88 * 0.021 *, and partner has a detectable mutation 0.04 * 0.88 *, and the fetus inherits both mutations 0.25).
Figure 2 examines the impact of an analytic specificity of 99.4% (described, alternatively, as a false-positive rate of 0.6%) by considering how many false-positive test results would occur in the same 10,000 non-Hispanic Caucasian women and their partners. Among the 58 women with false-positive test results (0.6% of the 9,600 women who are not carriers), two partners would be correctly identified as carriers (58 * 0.04 * 0.88 * 0.979). Among the 344 carrier women correctly identified (see previous paragraph), two partners would be expected to have a false-positive result [344 * (1–0.04) * 0.006]. Without confirmatory testing, these four couples would be misclassified as being at high risk and offered amniocentesis. In this population, there would be about 12 true-positive carrier couples (about 344 * 1/25 * 0.88 * 0.979). Such a high estimated proportion of couples with a false-positive result (4/16 or 25%) would be associated with fewer than the expected 1 in 4 offspring being affected with cystic fibrosis. This is not consistent with a summary analysis of published pilot trials.11 Among 55,000 pregnancies screened, 54 high risk couples were identified, and 18 homozygous fetuses were found (1 in 3). Two possible explanations for this discrepancy might be considered. First, laboratories participating in the trials may have performed confirmatory testing, thereby correcting most of the false-positive results before classifying couples as high risk. Second, analytic specificity may actually be higher than the 99.4% estimated from the ACMG/CAP survey. Three of the four false-positive results in that survey occurred during the first 2 years of proficiency testing (1996 and 1997). In practice, it will be important for screening laboratories to confirm the carrier status of couples classified as high risk. It will not be possible to identify false-positive couples based on the fetal genotype.

Flow diagram describing the impact of analytic sensitivity and analytic specificity of cystic fibrosis mutation testing in a population of 10,000 non-Hispanic Caucasian women and their partners undergoing prenatal screening. The analysis involves the same population and test characteristics as in Figure 1. Here, the added assumption is that 99.4% of the women with no detectable mutation will be correctly identified (analytic specificity). Thus 58 of 9,600 women with no mutation will receive a false-positive test result (0.6%). The box pointed to by the broken arrow contains relevant information derived from Figure 1.
Gaps in knowledge exist with regard to these preliminary estimates of analytic validity.11 Only overall estimates for analytic sensitivity and specificity are provided. It is likely that both method- and mutation-specific differences exist. For example, method-specific differences in analytic sensitivity are clearly demonstrated for the delI507 mutation. Some earlier methodologies had difficulty in distinguishing between G551D and R553X,5 and these mutations have been the source of problems in the ACMG/CAP program as well. Only 10 mutations have been challenged as part of proficiency testing (delF508, delI507, G542X, 621 + 1G>T, G85E, W1282X, G551D, R553X, 1717-1G>T, and R117H). Although these include the most common mutations, the majority of the 25 mutations in the panel recommended for prenatal screening have not yet been subjected to external proficiency testing. Lastly, it is possible that analytic performance will differ, depending on the numbers of mutations tested, even when the same methodology is employed. Standardized mutation panels using a higher number of mutations might be more robust because of automation or, conversely, the larger number of analytic steps might be more prone to errors.
A proficiency testing program (Survey FP) for maternal serum Down syndrome markers serves as one source for comparing error rates in non-DNA testing. In that survey (jointly sponsored by the Foundation for Blood Research and CAP),12 participating laboratories are asked to measure three biochemical markers, combine these measurements with a preassigned maternal age, and then calculate a Down syndrome risk. Five challenges are distributed three times each year. The proportion of laboratories with one or more outlying Down syndrome risk estimates on a given distribution is routinely reported to all participants each year. This proportion has remained relatively constant between 1998 and 2000 at about 5%. Assuming that the laboratory will have only one (or two) of the five risks classified as being an outlier, the actual error rate per sample distributed is about 1 or 2%. This is similar to the error rate for the ACMG/CAP MGL survey found in Table 1, suggesting that the analytic performance for this high-complexity DNA-based test is similar to non-DNA high-complexity laboratory tests.
References
National Institutes of Health. Secretary’s Advisory Committee on Genetic Testing. Enhancing the oversight of genetic tests: recommendations of the SACGT. Available at: http://www4.od.nih.gov/oba/sacgt.htm. Accessed July 2000.
American College of Obstetricians and Gynecologists, American College of Medical Genetics. Preconception, and prenatal carrier screening for cystic fibrosis. Clinical and laboratory guidelines: clinical and laboratory guidelines. Washington, DC, 2001.
Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ, Subcommittee on Cystic Fibrosis Screening, Accreditation of Genetic Services Committee, American College of Medical Genetics. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med 2001; 3: 149–154.
ACMG/CAP molecular genetics survey sets, 1996, 1997, 1998, 1999, 2000, 2001. Northfield, IL: College of American Pathologists.
Cuppens H, Cassiman JJ . A quality control study of CFTR mutation screening in 40 different European laboratories. Eur J Hum Genet 1995; 3: 235–245.
Dequeker E, Cassiman J . Genetic Proficiency testing in diagnostic laboratories: quality control is the message. Am J Hum Genet 2000; 67: A274.
Dequeker E, Cuppens H, Dodge J, Estivill X, Goossens M, Pignatti PF et al. Recommendations for quality improvement in genetic testing for cystic fibrosis. European Concerted Action of Cystic Fibrosis. Eur J Hum Genet 2000; 8: s2–s24.
Kazazian HH for the Cystic Fibrosis Genetic Analysis Consortium. Population variation of common cystic fibrosis mutations. Hum Mutat 1994; 4: 167–177.
Palomaki GE, Haddow JE, Bradley LA, FitzSimmons SC . An updated assessment of cystic fibrosis mutation frequencies in non-Hispanic Caucasians. Genet Med 2002; 4: 90–94.
Cunningham S, Marshall T . Influence of five years of antenatal screening on the paediatric cystic fibrosis population in one region. Arch Dis Child 1998; 78: 345–348.
Haddow JE, Palomaki GE, editors. Population-based prenatal screening for cystic fibrosis via carrier testing: ACCE review. Available at: http://www.fbr.org/projects/acce-cdc/acce_doc.html. Accessed October 3, 2002.
FBR/CAP maternal serum survey sets, 1999 and 2000. Northfield, IL: College of American Pathologists, 1998.
Acknowledgements
Support was provided by a cooperative agreement (UR3/CCU319352) with the Centers for Disease Control and Prevention, Office of Genomics and Disease Prevention. The data source for this analysis was designed, collected, and reported by the Biochemical and Molecular Genetics Resource Committee of the College of American Pathologists and the American College of Medical Genetics. The authors thank the Committee members for their comments and suggestions.
Author information
Authors and Affiliations
Additional information
A complete listing of the sample challenges, the types of errors, and adjustments made during the analysis is available via the ArticlePlus feature at the Genetics in Medicine Web site, http://www.geneticsinmedicine.org.
Rights and permissions
About this article
Cite this article
Palomaki, G., Bradley, L., Richards, C. et al. Analytic validity of cystic fibrosis testing: A preliminary estimate. Genet Med 5, 15–20 (2003). https://doi.org/10.1097/00125817-200301000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200301000-00003
Keywords
This article is cited by
-
Chapter 11: Challenges in and Principles for Conducting Systematic Reviews of Genetic Tests used as Predictive Indicators
Journal of General Internal Medicine (2012)
