
Abstract
Purpose: To report on the use of fluorescence in situ hybridization (FISH) and dosage-sensitive Southern blot analysis in the molecular diagnosis of patients with Saethre-Chotzen syndrome.
Methods: FISH and dosage-sensitive Southern blot analysis utilizing TWIST gene probes were performed on patients with Saethre-Chotzen syndrome but without an identifiable TWIST sequence variation.
Results: Four unrelated patients with a deletion of the TWIST gene were identified by Southern blot; one of them had a complex chromosomal rearrangement involving 7p21 and no apparent deletion by FISH, suggesting a smaller deletion in the region including the TWIST gene. A fifth patient had an abnormal TWIST gene fragment on Southern blot analysis that segregated with the disease in the family; FISH was normal in this patient, suggesting a partial deletion or rearrangement in or near the gene.
Conclusion: FISH and dosage-sensitive Southern blot analysis are useful diagnostic tools in Saethre-Chotzen syndrome without TWIST sequence variation.
Similar content being viewed by others


Main
Saethre-Chotzen syndrome (OMIM 101400) is a relatively common craniosynostosis syndrome, named after the authors of its first reports.1–3 Numerous familial cases have been reported, demonstrating the autosomal dominant inheritance as well as wide intrafamilial variability. The classical presentation includes bicoronal synostosis with the resulting brachycephaly or acrocephaly; but other sutures may be affected or there may be facial asymmetry rather than overt craniosynostosis. Frequently noted findings include a low anterior hairline, ptosis, hypertelorism, small and apparently posteriorly rotated ears with a prominent crus, partial cutaneous syndactyly of hands and feet, and hallux valgus. In addition, lacrimal duct stenosis, cleft palate, radioulnar synostosis, vertebral abnormalities, broad thumbs, and a partial duplication of the first toe are also seen in some patients with Saethre-Chotzen syndrome.4 Because of this variability in physical findings, there is significant phenotypic overlap with other craniosynostosis conditions, such as the fibroblast growth factor receptor 3 (FGFR3) P250R mutation, making molecular confirmation of the presumed clinical diagnosis an important prerequisite for counseling of patients and family members.5,6 Associated problems, such as learning difficulties, may be more or less likely, depending on the molecular etiology. Identification of an underlying mutation in a parent allows for early prenatal diagnosis by molecular techniques.
The locus for Saethre-Chotzen syndrome on 7p21 was identified through linkage studies and breakpoint analysis of affected individuals carrying translocations.7,8 Later, the human TWIST gene, encoding a helix-loop-helix transcription factor, was mapped to this region. It was an attractive candidate gene because heterozygous Twist null mice showed skull and limb abnormalities reminiscent of the findings in humans with Saethre-Chotzen syndrome.9 Subsequently, mutations in the human TWIST gene were identified in the majority of patients with Saethre-Chotzen syndrome.9,10 Point mutations throughout the TWIST gene have been found to cause Saethre-Chotzen syndrome.11 Since many of these are nonsense mutations, haploinsufficiency for the functional gene product was hypothesized as the disease-causing mechanism. Haploinsufficiency may also be caused by deletion of one allele. For this reason, Johnson et al.12 hypothesized that gene deletions might account for cases in which no point mutation was identified. They found complete gene deletions in 4 patients and 1 first-degree relative using a combination of FISH, polymerase chain reaction (PCR) haplotype, and Southern analysis, thus demonstrating that gene deletions cause a significant proportion of Saethre-Chotzen syndrome cases.
We have developed a comprehensive diagnostic assay for the molecular diagnosis of Saethre-Chotzen syndrome, consisting of DNA sequence, dosage-sensitive Southern blot, and molecular cytogenetic analysis by FISH. Using this combined approach, we were able to demonstrate complete gene deletions in four patients and a partial gene deletion or rearrangement in a fifth case.
PATIENTS AND METHODS
All probands were diagnosed with Saethre-Chotzen syndrome based on their clinical findings (Table 1). The family members of patient 5 were diagnosed with Saethre-Chotzen syndrome based on subtle clinical findings without overt craniosynostosis (Table 1).
Patient 1
This male was born after an uneventful pregnancy at 36 weeks gestational age by spontaneous vaginal delivery. His 27-year-old mother had lost one pregnancy by early spontaneous abortion. His father was 33 years old. The nonconsanguineous parents were in good health and neither had a family history of craniosynostosis. The proband was found to have bicoronal synostosis at birth, which was surgically repaired at age 1 month and again at age 1 year. Physical findings were consistent with a diagnosis of Saethre-Chotzen syndrome (Table 1). Except for feeding difficulties in early infancy, there were no other medical problems. Gross motor development was delayed with sitting at 7 months and walking at 18 months of age. At his current age of 9 years, he requires special help in school and has a full-scale IQ of 73.
On chromosome analysis, a marker chromosome was seen in all cells; there was no other abnormality noted. Parental studies revealed that the marker was of paternal origin, as the nondysmorphic father carried the same marker in all cells examined. Comparative genomic hybridization showed the marker to be derived from chromosome 14.
Patient 2
This female was born by spontaneous vaginal delivery at full term as the third child of a 30-year-old mother and a 31-year-old father. Her mother had one early spontaneous abortion. The patient's father was reportedly in good health until he died suddenly of undetermined cause at age 43 years. Neither parent had signs of craniosynostosis on physical examination. The patient's siblings are reportedly in good health. The proband was diagnosed with bicoronal synostosis in early infancy, and the diagnosis of Saethre-Chotzen syndrome was made based on the additional physical findings (Table 1). Surgeries for craniosynostosis repair and associated facial malformations were performed at 6 months, 7 years, and 13 years. A pharyngoplasty for submucous cleft palate was performed at age 11 years. She had generalized developmental delays and, at the current age of 14 years, requires special education. Her full-scale IQ is reportedly 50; and she has attention deficit disorder. Chromosomal analysis with G-banding demonstrated a 46, XX karyotype.
Patient 3
This patient was the female full-term product of an uncomplicated pregnancy, born to a 23-year-old mother and a 34-year-old father. Both parents were in good health and had no family history of craniosynostosis, but her mother reportedly had four early spontaneous abortions. Craniosynostosis was diagnosed in early infancy and repaired surgically at 6 months and 8 years of age. Ptosis repair was performed at 3 years of age. The diagnosis of Saethre-Chotzen syndrome was made clinically (Table 1). She had generalized developmental delays, is considered neurologically impaired, and requires special education.
Patient 4
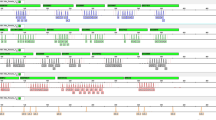
A male infant was delivered at term after a pregnancy complicated by fetal ventriculomegaly noted on ultrasound at 5 months gestation. His 37-year-old mother and 43-year-old father are nonconsanguineous and have three other healthy children, his mother had three spontaneous abortions. Bicoronal synostosis was noted, and Saethre-Chotzen syndrome was diagnosed based on the additional clinical findings (Table 1). The patient also had unilateral cryptorchidism, a VSD, and an ASD. Chromosomal analysis revealed a complex chromosomal rearrangement, involving a total of six breakpoints on four chromosomes. The karyotype was designated as 46, XY, der(1)t(1;2;7;18)(q31.3;q24.3;p21.1;p11.3), der(2)t(1;2;7;18), der(7)t(1;2;7;18)t(7;18)(q21.2;q21.31), der(18)t(1;2;7;18)t(7;18). The partial composite karyotype (Fig. 1 A) shows the four derivative chromosomes with their normal homologues. The complex rearrangement appears to have resulted from two balanced de novo translocations. One was a complex, four-break chromosomal rearrangement involving the long arm of chromosome 1, the long arm of chromosome 2, the short arm of 7, and the short arm of 18. The other involved the long arms of the same chromosome 7 and 18 homologues (Fig. 1B). The breakpoint in the short arm of chromosome 7 appears to lie very near the 7p15/7p21 border and could not be unambiguously assigned by cytogenetic analysis. Results of successive dual color FISH analyses were consistent with the interpretation from routine cytogenetic analysis. Parental karyotypes were normal, as was dosage-sensitive Southern analysis for a TWIST gene deletion.

A: Patient 4's partial composite karyotype shows the four derivative chromosomes with their normal homologues. B: Schematic representation of patient 4's derivative chromosomes, indicating the translocation breakpoints by horizontal bars. The band assignment is given on the respective normal homologues.
Patient 5
This male was delivered at full term after an uneventful pregnancy by repeat cesarean section. Ptosis was noted in early infancy, and three surgical procedures were performed for this reason. At 2 years of age, bicoronal synostosis was seen on radiographs, and subsequently the sagittal and lambdoid sutures were also noted to be affected. He was diagnosed with Saethre-Chotzen syndrome (Table 1). At age 28 months, his motor development was mildly delayed and language development was delayed. Chromosomal analysis showed a normal 46, XY karyotype.
Patient 5A
This patient is the brother of patient 5. He does not have craniosynostosis but was noted to have subtle signs of Saethre-Chotzen syndrome (Table 1), including tear duct stenosis requiring surgical repair, and partial duplication of the first toes. His development is thought to be appropriate for his current age of 7 years. Previously, chromosome analysis of the patient displayed a normal result.
Patient 5B
The mother of patients 5 and 5A is currently 31 years old. She has not been diagnosed with craniosynostosis, but has subtle findings consistent with Saethre-Chotzen syndrome (Table 1), including brachydactyly with broad thumbs and first toes. She reported that her brother, mother, and her deceased maternal uncle shared these findings. Her mother's cousin and two of their grandchildren reportedly required surgical repair for craniosynostosis.
TWIST sequence analysis
Sequence analysis was performed using standard techniques as described previously.13
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) of metaphase chromosomes from peripheral blood was performed using standard techniques. Cosmid IIIA9,14 a 45-kb cosmid spanning the complete TWIST gene and >20 kb of 5′ and 3′ flanking sequences was used as FISH probe. The cosmid was labeled with digoxigenin and detected with rhodamine. DAPI staining of the same metaphase image allowed for identification of the chromosomes 7 by the G-band pattern, and presence or absence of the FISH signal was assessed.
Dosage-sensitive Southern blot analysis
Genomic DNA was extracted using a commercially available kit (Gentra systems, Inc., Minneapolis, MN). Restriction enzyme digestion of 5 μg of genomic DNA with Eco RI was followed by electrophoresis on an 1% agarose gel and transfer of the DNA to a nitrocellulose membrane (NitroPure, Microsystems, Inc.) by capillary blotting. The blot was hybridized to a 32P-labeled probe generated by PCR amplification of the TWIST coding sequence, and a control probe specific for the beta globin gene. Two different probes were used for the TWIST gene in sequentially performed studies. Both probes were generated by PCR amplification of genomic DNA: One probe was a 499-bp fragment complementary to sequences encoding amino acids 99 to 202 plus 187 bp of intron sequence. The second probe was a 591-bp fragment complementary to the last 132 bp of the intron plus 459 bp of the 3′ UTR. Both probes yielded identical results on all Southern blots. After a final wash with 0.2X SSC + 0.1% SDS at 65°C, the filters were exposed to x-ray film at −70°C. Evaluation of gene dosage was performed by visual comparison of the signal intensities of fragments hybridizing to the TWIST and the beta globin probes.
RESULTS
Sequence analysis of the TWIST gene performed on all index patients showed only the normal DNA sequence. In order to identify a molecular basis for the clinical diagnosis of Saethre-Chotzen syndrome, additional studies were performed as samples were available (Table 2).
Dosage-sensitive Southern blot analysis revealed that patients 1, 2, 3, and 4 were heterozygous for deletion of the TWIST gene. With equal loading of DNA samples as assessed by equal intensities of the beta globin band in each lane, decreased intensity of the 7.7-kb Twist fragment indicates heterozygosity for a TWIST gene deletion. Two of the patients (1 and 2) were also shown to be deleted for one copy of the gene by FISH. Patient 3 was unavailable for FISH. The fourth patient displayed two hybridization signals by FISH, but one signal was on a translocated segment of chromosome 7, on the der(7)t(1;2;7;18)t(7;18)(q21.2;q21.31) at the appropriate position.
Dosage-sensitive Southern blot analysis on patient 5 showed one fragment of normal size (7.7 kb), with decreased intensity, and an additional band of 10 kb (Fig. 2, lanes 5, 7, and 8) indicating a partial deletion or rearrangement of the DNA fragment containing the TWIST gene. Abnormal size bands were also observed on Southern blot analysis of genomic DNA digested with Sac I and Nco I (data not shown).

Dosage-sensitive Southern blot of DNA from family 5. Blot of Eco RI-digested genomic DNA from affected and unaffected family members was hybridized with 32P-labeled probes specific for the TWIST and the beta globin genes. An abnormally migrating band was observed in lanes containing DNA from the affected mother (•) and sons (▪). C, control DNA; D, DNA from a patient with a known deletion of the TWIST gene.
Samples were obtained on affected and unaffected members of family 5. Southern blot analysis of Eco RI-digested DNA demonstrated that the abnormal band segregated with the disease in this family (Fig. 2). Segregation analysis in this family is, thus, consistent with a partial deletion or rearrangement in or around the TWIST gene.
FISH analysis of the mother (patient 5B) showed two hybridization signals.
DISCUSSION
We performed dosage-sensitive Southern blot analysis on five probands with Saethre-Chotzen syndrome in whom no mutation was identified on TWIST sequence analysis. Patients 1, 2, and 3 were heterozygous for a gene deletion by this approach. FISH analysis confirmed this deletion in patients 1 and 2. Patient 3 was unavailable for FISH. Parental studies in patient 1 showed that this deletion occurred de novo in the patient.
In patient 4, a gene deletion was evident by the Southern blot assay, while no deletion was detected by FISH. However, one hybridization signal was located on a derivative chromosome, and the patient's karyotype shows a complex chromosomal rearrangement. It is possible that the TWIST gene was deleted as a result of the chromosomal rearrangement. Since the cosmid used for FISH analysis is 45 kb in size and contains <1 kb of TWIST coding sequence, it is possible that small deletions or even complete deletions of the TWIST coding region might be missed by FISH if sufficient genomic sequence remains to which the cosmid probe can hybridize. In this situation, the Southern blot assay, using the TWIST coding region as probe, appears to be a more sensitive indicator of TWIST gene deletions.
The Southern blot analysis in patient 5 showed a second band in addition to the normal size band. Additional bands were also seen after digestion with two other restriction enzymes, indicating that the abnormal bands were not likely to be due to a restriction fragment length polymorphism (RFLP). These data are suggestive of a submicroscopic rearrangement, such as a deletion, insertion, or inversion, in the region of the TWIST gene. Restriction enzyme digestion of control DNA with Nco I yields a band of 2.8 kb containing the TWIST gene. In the patient, an additional band of increased size was seen after Nco I digestion, suggesting that the rearrangement or deletion is in close proximity to the TWIST coding sequence. Although it is not clear whether the TWIST gene itself is interrupted in this patient, such a defect could, by its proximity to the gene, affect gene expression and, thus, lead to haploinsufficiency. Studies of the TWIST gene expression in this patient and patient 4 are under way.
Johnson et al.,12 used the same cosmid as in this study for their FISH analysis and reported a visible deletion in three probands with deletions ranging in size from 3.5 to >11 Mb, whereas a smaller deletion of 2.9 kb was not visualized by FISH. Of note is that the patients with the larger deletions had learning difficulties, whereas the proband and her relative with the smaller deletion were intellectually normal. Our patients 1 and 2 had deletions visualized on FISH, and both show significant learning problems. Patient 4 is too young to assess, and his development may be compromised by additional abnormalities due to the complex chromosomal rearrangement. Patient 5 has language delay. His brother (5A) and his mother (5B) carry the same molecular defect, and both have normal intellectual development, as do other family members who are suspected of carrying the mutation based on clinical reports. FISH performed on patient 5B had normal results, indicating retention of significant portions of DNA complementary to the cosmid probe. In this family, the mutation in the vicinity of the TWIST gene apparently segregates through multiple generations, suggesting little or no intellectual deficits associated with it. Thus, our patients may, in combination with those reported by Johnson et al., suggest that patients with a TWIST deletion large enough to be visible by FISH are prone to significant learning difficulties. Currently, the number of patients with smaller deletions, identified by Southern blot but not visualized on FISH, is too small and too heterogeneous to permit conclusions regarding intellectual development. Patients with large deletions may become valuable for the identification of genes involved in brain function.
The identification of underlying gene mutations has greatly enhanced our understanding of craniosynostosis disorders.15 The diagnosis of Saethre-Chotzen syndrome can often be made clinically in familial cases, when affected family members are available for examination. In other instances, when the phenotype is not distinctive, the identification of an underlying TWIST gene mutation is crucial for the diagnosis.13 This information can then be used to identify mutation carriers and to perform prenatal diagnosis. While TWIST sequence analysis is often successful in identifying an abnormality, some patients with Saethre-Chotzen syndrome do not show a sequence variation. These patients may have a submicroscopic 7p21 deletion, including the TWIST coding sequence.
In order to assess the relative frequency of TWIST gene deletions compared to point mutations, we reviewed the patient population followed in our institution. We identified a gene deletion in 3 (patients 1–3 in this report) and a point mutation in 10 probands diagnosed with Saethre-Chotzen syndrome, giving a relative frequency of 3/13 or 23%. Johnson et al.12 found a gene deletion in three and a point mutation in five patients clinically diagnosed with Saethre-Chotzen syndrome, yielding a relative frequency of 3/8 or 37.5%. Combining the cases from these studies, the relative frequency is 6/21 or 28.5%, suggesting that a gene deletion is the cause of Saethre-Chotzen syndrome in a significant percentage of cases.
Analysis for a TWIST gene deletion should be included as part of a molecular assay for Saethre-Chotzen syndrome. We perform this molecular analysis as outlined in the flow diagram (Fig. 3). When the diagnosis of Saethre-Chotzen syndrome is considered, we recommend that TWIST sequence analysis be performed, as the majority of patients will have an identifiable sequence variation. If no sequence variation is detected, a quantitative Southern blot is recommended. If a deletion is identified by Southern blot, FISH analysis may provide additional information regarding the deletion size and the associated risk for learning differences. In cases with an identified TWIST mutation, analysis of first-degree relatives may be considered because of the known intrafamilial variability of Saethre-Chotzen syndrome.4,13 If no mutation in TWIST is detected, we suggest testing for the P250R mutation in FGFR3, since patients with this mutation may not always be clinically distinguishable from those with Saethre-Chotzen syndrome.6 Analysis for the FGFR3 mutation may be included at any stage of the molecular studies. Because of the phenotypic overlap, it may not be helpful to attempt prioritization. If the patient has additional findings not typical for Saethre-Chotzen syndrome, we may also recommend a chromosome analysis with particular attention to 7p21, the location of the TWIST gene, because a translocation or a complex chromosomal rearrangements may be present.

Flow-diagram for molecular and cytogenetic evaluation of patients with Saethre-Chotzen syndrome.
In summary, we successfully incorporated dosage-sensitive Southern blot analysis and FISH in the diagnostic evaluation for patients with Saethre-Chotzen syndrome. Southern blot analysis can be performed on the same blood sample submitted for sequence analysis and has a higher detection rate for deletions than FISH. However, FISH can provide valuable information regarding deletion size and possible associated developmental problems. These assays can elucidate the cause of Saethre-Chotzen syndrome in cases without identifiable TWIST sequence variation and add valuable information for counseling of the family. For example, in an infant or young child found to be at risk for developmental delay, early intervention can be instituted before significant delays occur. In addition, these cases may further our understanding of the molecular mechanisms causing Saethre-Chotzen syndrome and the learning difficulties noted in deletion carriers.
References
OMIM (Online Mendelian Inheritance in Men) Baltimore. Johns Hopkins University, Center for Medical Genetics, 2000. http://www4.ncbi.nlm.nih.gov/Omim/.
Saethre H . Ein Beitrag zum Turmschaedelproblem (Pathogenese, Erblichkeit und Symptomologie). Dtsch Z Nervenheilkd 1931; 117: 533–555.
Chotzen F . Eine eigenartige famililaere Entwicklungsstoerung (Akrocephalosyndactylie, Dysostosis craniofacialis und Hypertelorismus). Monatschr Kinderheilkd 1932; 55: 97–.
Pantke OA, Cohen MM, Witkop Jr CJ, Feingold M, Schaumann B, Pantke HC, Gorlin RJ . The Saethre-Chotzen syndrome. Birth Defects Orig Artic Ser 1975; 11: 190–225.
Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, Markowitz RI, Robin NH, Nwokoro N, Mulvihill JJ, Losken HW, Mulliken JB, Guttmacher AE, Wilroy RS, Clarke LA, Hollway G, Ades LC, Haan EA, Mulley JC, Cohen MM, Bellus GA, Francomano CA, Moloney DM, Wall SA, Wilkie AO, Zackai EH . A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997; 60: 555–564.
Paznekas WA, Cunningham ML, Howard TD, Korf BR, Lipson MH, Grix AW, Feingold M, Goldberg R, Borochowitz Z, Aleck K, Mulliken J, Yin M, Jabs EW . Genetic heterogeneity of Saethre-Chotzen syndrome, due to TWIST and FGFR mutations. Am J Hum Genet 1998; 62: 1370–1380.
Brueton LA, van Herwerden L, Chotai KA, Winter RM . The mapping of a gene for craniosynostosis: evidence for linkage of the Saethre-Chotzen syndrome to distal chromosome 7p. J Med Genet 1992; 29: 681–685.
van Herwerden L, Rose CS, Reardon W, Brueton LA, Weissenbach J, Malcolm S, Winter RM . Evidence for locus heterogeneity in acrocephalosyndactyly: a refined localization for the Saethre-Chotzen syndrome locus on distal chromosome 7p-and exclusion of Jackson-Weiss syndrome from craniosynostosis loci on 7p and 5q. Am J Hum Genet 1994; 54: 669–674.
el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J . Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet 1997; 15: 42–46.
Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW . Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet 1997; 15: 36–41.
Gripp KW, Zackai EH, Stolle CA . Mutations in the human TWIST gene. Hum Mutat 2000; 15: 150–155.
Johnson D, Horsley SW, Moloney DM, Oldridge M, Twigg SR, Walsh S, Barrow M, Njolstad PR, Kunz J, Ashworth GJ, Wall SA, Kearney L, Wilkie AO . A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am J Hum Genet 1998; 63: 1282–1293.
Gripp KW, Stolle CA, Celle L, McDonald-McGinn DM, Whitaker LA, Zackai EH . TWIST gene mutation in a patient with radial aplasia and craniosynostosis: further evidence for heterogeneity of Baller-Gerold syndrome. Am J Med Genet 1999; 82: 170–176.
Krebs I, Weis I, Hudler M, Rommens JM, Roth H, Scherer SW, Tsui LC, Fuchtbauer EM, Grzeschik KH, Tsuji K, Kunz J . Translocation breakpoint maps to 5 kb 3′ from TWIST in a patient affected with Saethre-Chotzen syndrome. Hum Mol Genet 1997; 6: 1079–1086.
Wilkie AO . Craniosynostosis: genes and mechanisms. Hum Mol Genet 1997; 6: 1647–1656.
Acknowledgements
We thank Dr. Brynn Levy for performing comparative genomic hybridization on patient 1. Dr. Juergen Kunz kindly provided the cosmid for FISH. This investigation was supported in part by the Public Health Services Research Grant M01-RR00240 from the National Institutes of Health.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gripp, K., Kasparcova, V., McDonald-McGinn, D. et al. A diagnostic approach to identifying submicroscopic 7p21 deletions in Saethre-Chotzen syndrome: Fluorescence in situ hybridization and dosage-sensitive Southern blot analysis. Genet Med 3, 102–108 (2001). https://doi.org/10.1097/00125817-200103000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200103000-00003
Keywords
This article is cited by
-
Destabilization of the TWIST1/E12 complex dimerization following the R154P point-mutation of TWIST1: an in silico approach
BMC Structural Biology (2018)
-
Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses
Child's Nervous System (2012)
