
Abstract
Costello syndrome (OMIM# 218040) is a distinctive rare multisystem disorder comprising a characteristic coarse facial appearance, intellectual disabilities, and tumor predisposition. Although the diagnosis can be suspected clinically, confirmation requires identification of a heterozygous mutation in the proto-oncogene HRAS. In contrast to somatic oncogenic mutations in neoplasia, the Costello syndrome changes are typically introduced in the paternal germline. The predicted amino acid substitutions allow for constitutive or prolonged activation of the HRAS protein, resulting in dysregulation of the Ras/mitogen activated protein kinase pathway. Dysregulation of this signaling pathway is the disease mechanism shared among Costello syndrome and other rasopathies, including neurofibromatosis type 1, Noonan syndrome, cardio-facio-cutaneous syndrome, and Legius syndrome. The Ras/mitogen activated protein kinase pathway governs cell proliferation and differentiation, and its dysregulation affects cardiac and brain development, accounting for the significant overlap in physical and developmental differences and common medical problems among rasopathies. Unlike the genetically heterogeneous Noonan syndrome and cardio-facio-cutaneous syndrome, Costello syndrome is caused by HRAS mutations only. Patients, clinicians, and researchers may benefit from a multidisciplinary “rasopathy clinic,” which serves patients with more common conditions such as Noonan syndrome and neurofibromatosis and those affected by rare conditions such as Costello syndrome.
Genet Med 2012:14(3):285–292
Similar content being viewed by others


Introduction
Costello syndrome (OMIM# 218040) is a rare disorder first described 40 years ago by a New Zealand pediatrician1,2 based on its distinctive phenotype. Characteristic findings include coarse facial features, intellectual disability, cardiac problems (typically cardiac hypertrophy, less commonly, atrial tachycardia, or pulmonic stenosis), and severe failure-to-thrive with short stature.3 The predisposition to neoplasia encompasses a spectrum from benign wart-like skin lesions termed papillomata to malignant tumors including rhabdomyosarcoma, neuroblastoma, and transitional cell carcinoma of the bladder.4 The identification of activating heterozygous germline mutations in the proto-oncogene HRAS in individuals with Costello syndrome5 provided a rationale for the increased malignancy risk. Further, additional gene changes affecting other components of the Ras/mitogen activated protein kinase (MAPK) pathway were found to result in the clinically related conditions—Noonan syndrome and cardio-facio-cutaneous (CFC) syndrome. These conditions are now collectively referred to as rasopathies,6 and the improved understanding of the Ras/MAPK pathway’s primary role in embryologic development has shifted the focus from its previously well studied role in carcinogenesis. In contrast to the molecular heterogeneity of CFC and Noonan syndrome, Costello syndrome results from mutations in a single gene. Its gene product plays a critical role within the signaling pathway as it alternates between its active, signal transmitting and its inactive conformation. The activating point mutations in Costello syndrome shift the balance toward the active, signal transmitting state, resulting in dysregulation of the signaling pathway. Costello syndrome can be considered a model disorder for rasopathies, and further phenotype/genotype studies are emerging.
Discussion
Clinical diagnosis
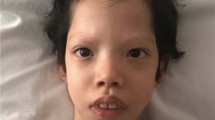
Costello syndrome can be diagnosed clinically with the greatest confidence in the older child. Formal diagnostic criteria for Costello syndrome have not been developed, but consensus guidelines developed by experts have been published.7 No single feature is unique for Costello syndrome, although the constellation of several creates the characteristic facial appearance with curly or fine hair, prominent epicanthal folds, long eyelashes, full nasal tip, fleshy ear lobes, and a wide mouth with full lips. When supplemented by the distinctive hand position (severe ulnar deviation), diffuse hyperpigmentation, hoarse voice and generally, friendly outgoing personality, the memorable phenotype is complete ( Table 1 ) ( Figure 1a,b ). Clinicians should view these guidelines in the context of the natural history described later, as physical manifestations evolve in an age-dependent manner.

Typical facial features of Costello syndrome. (a) An 8-year-old white boy and (b) an almost 11-year-old Hispanic girl. The features are remarkably consistent regardless of sex or ethnic background. Note the high broad forehead, low-set ears with thickened lobules, prominent epicanthal folds, full cheeks, wide mouth, full lips, and radiant smiles which are typical for the pleasant personality of Costello syndrome. The appearance of the girl can be compared with an earlier photograph taken 1 year ago (Figure 3D in ref. 8). There are some changes including thinning of the frizzy hair, wrinkling of the skin, and wearing of corrective lenses. We thank the mothers of these patients for contributing their personal photographs. Permission to publish these photographs was given by the mothers.
Molecular genetic testing
A suspected clinical diagnosis of Costello syndrome is confirmed through the identification of a specific germline mutation in the proto-oncogene HRAS, which is the only gene associated with Costello syndrome according to a review by expert clinicians.5,9 Sequence analysis of HRAS exon 2 (the first coding exon) detects missense mutations in 80–90% of individuals tested.5,10,11,12 If no mutation is identified in exon 2, all other coding exons and flanking intronic regions should be sequenced, as rare mutations have been seen in other exons.
Failure to identify an HRAS mutation in an individual with a phenotype thought to be typical for Costello syndrome can result from either (1) the rare occurrence of low level of somatic mosaicism for the HRAS disease-causing mutation in the tested tissue11,12,13,14 or (2) the more common presence of a mutation in another gene, consistent with a diagnosis of CFC syndrome15,16 or another disorder of the Ras/MAPK pathway.17 In the early series, the 10–15% of individuals suspected of having Costello syndrome who lacked an HRAS mutation were later found to have CFC syndrome.15,16
Molecular genetic pathogenesis
HRAS is a well-known proto-oncogene, and aberrant activation is often found in sporadic somatic tumors; it is thus not surprising to see the increased cancer incidence in individuals with a germline HRAS mutation. The work performed by Kerr et al.18 showing loss of heterozygosity for 11p15.5 in rhabdomyosarcoma from individuals with Costello syndrome suggests that loss of the wild-type allele is the second hit in tumor development. This theory is supported by the loss of the wild-type allele in a rhabdomyosarcoma demonstrated by Estep et al.10 and the monoallelic expression in a tumor, but not in fibroblasts, reported by Aoki et al.5
HRAS consists of six exons. Five exons2,3,4,5,6 code for a protein of 189 amino acids with a molecular weight of 21 kd (p21). Alternative splicing, excluding residues 152–165, gives rise to a protein of 170 amino acids. Somatic mutation hotspots are bases encoding the glycines in Positions 12 and 13 and the glutamine in Position 61. Missense mutations at these positions lead to increased activity of the gene product. As the germline mutations in Costello syndrome affect similar codons, it can be inferred that they have a similar effect on the gene product. Functional studies performed on patient-derived fibroblasts confirmed aberrant downstream signaling.19
Germline mutations leading to nucleotide substitutions and the consequent amino acid substitutions of the glycine residue at Positions 12 or 13 are typical in Costello syndrome.20 A review of 81 unrelated individuals5,11,12 shows the nucleotide substitution c.34G>A, resulting in p.Gly12Ser amino acid change, to be the most common (65/81 or 80%). The c.35G>C nucleotide resulting in p.Gly12Ala was seen in seven individuals (9%). Estep et al.10 reported the same two mutations and aggregate clinical data on 33 individuals. In this study11 and others conducted by colleagues, we have avoided duplicated reporting of this rare syndrome.21 Recently, 12 individuals with p.Gly13Cys change were identified,8 making this the most common amino acid change affecting the glycine in Position 13. Other mutations ( Table 2 ) were seen in one or two patients each.5,10,11,12,22,23,24,25
Natural history
Females and males are equally affected. The phenotype evolves over time, and although Costello syndrome can theoretically be recognized in the fetus, it is usually diagnosed in the young child. Older individuals often display features suggestive of premature aging. Prenatally, increased nuchal thickness, polyhydramnios (>90%), characteristic ulnar deviation of the wrists, and short humeri, and femurs can be seen on ultrasonography.26,27 Because most features of the fetal phenotype are not unique (such as ventriculomegaly and macrocephaly), and as Costello syndrome is rare, the diagnosis is often not considered prenatally. Cardiac hypertrophy has not been reported, but fetal atrial tachycardia has been detected in at least five fetuses subsequently diagnosed with Costello syndrome, and it increases the index of suspicion for this diagnosis.26 Increased birth weight and head circumference (often >50th percentile) for gestational age can lead to the misclassification of Costello syndrome as “macrosomia.” Hypoglycemia is common in neonates. Failure to thrive and severe feeding difficulties are usually severe and almost universal.
Characteristic physical findings include a relatively high forehead, low nasal bridge, epicanthal folds, prominent lips and a wide mouth, ulnar deviation of wrists and fingers, loose- appearing skin with deep palmar and plantar creases, and cryptorchidism. In infancy, severe feeding difficulties may lead to a marasmic appearance. Pyloric stenosis occurs more commonly than in the general population.24 Most infants display hypotonia, irritability, developmental delay, and nystagmus with delayed visual maturation improving with age. Hypotonia may be severe and suggest myopathy.22 Relative macrocephaly is typical and likely reflects brain overgrowth.28 Progressive postnatal cerebellar enlargement results in cerebellar tonsillar herniation, which can progress to a Chiari malformation as seen in 32% of individuals in one series, with 25% developing syringomyelia.28 Neurosurgical interventions included posterior fossa decompression (32%), shunt placement or ventriculostomy (25%), and tethered cord release (7%), reflecting the significant morbidity associated with brain overgrowth.28 Nystagmus is common and may improve over time, although its underlying cause has not been determined. Although nystagmus does not require medical treatment, it is important to recognize it as a common finding in Costello syndrome, rather than an unrelated symptom.
In childhood, individuals are able to take oral feeds beginning between age 2 and 4 years. The first acceptable tastes are often strong (e.g., ketchup). The onset of speech often coincides with the willingness to feed orally. Short stature is universal, delayed bone age is common,29 and testing may show partial or complete growth hormone (GH) deficiency.11
Cardiovascular abnormalities are typically present in infancy or early childhood, but may be recognized at any age. Based on a recent analysis of 61 study and 85 previously reported patients with HRAS mutation analysis, 87% of individuals with Costello syndrome have some type of a cardiovascular abnormality, increased from a prior report based on clinically diagnosed patients (60%).30 A congenital heart defect was present in 44%, usually nonprogressive valvar pulmonic stenosis, rarely atrial septal defect. Cardiac hypertrophy was noted in 61%; the term hypertrophic cardiomyopathy (HCM) was used because there was typical subaortic septal hypertrophy associated with pathologic myocardial disarray (in 70% of specimens studied). In addition to the rare lethal severe form, HCM was chronic or progressive in 37%, stabilized in 27%, decreased (resolved) in 15%; and outcome was unavailable in 22%. Atrial tachycardia occurred in 48%, characteristically as multifocal atrial tachycardia (also known as chaotic atrial rhythm). These nonreentrant atrial tachycardias are generally self-limited but may persist or worsen in approximately one fourth. Mild-moderate aortic dilation occurs in approximately 5% and was unassociated with bicuspid aortic valve.
Although the cardiovascular natural history of Costello syndrome continues to be delineated, this recent data can be used to create several “cardiac phenotypes.” A few neonates present with very severe HCM can be lethal if untreated. In other infants, progressively severe HCM and/or severe multifocal atrial tachycardia can lead to death in the first 2 years of life. However, most children with Costello syndrome associated with HCM have mild to moderate involvement. Of great interest are the few with moderate to severe involvement who appear to have remodeling, which gives the impression of “normalizing.” Multifocal atrial tachycardia and ectopic atrial tachycardia may be concerning but are usually self-limited with treatment. Pulmonic valve stenosis is usually mild to moderate and infrequently requires surgery or interventional catheterization. Older individuals with moderate HCM and new onset arrhythmia (atrial and ventricular) represent the greatest challenge and do not constitute a predictable outcome “phenotype” until more information is obtained.
The possibility of a congenital myopathy in Costello syndrome was hypothesized based on the observation of four patients with severe hypotonia, three of whom had less common HRAS mutations.22 On muscle biopsy, there were excess muscle spindles, leading to the description “congenital myopathy with excess of muscle spindles.” The analysis of skeletal muscle pathology was extended by Tidyman et al.31 who studied additional pathology reports and muscle biopsies. They found abnormal muscle fiber size and variability, type 2 fiber predominance, but without severe structural histopathology and no excess of muscle spindles. Because Costello syndrome mutations inhibited myoblast differentiation, they agreed that there was evidence of a true myopathy related to an inherent dysregulation of skeletal myogenesis.31
Papillomata, absent in infancy, appear in young children. Acanthosis nigricans, thick calluses and toenails, and strong body odor are common. Developmental delay or intellectual disability is present in all individuals.32,33,34,35 Electroencephalogram abnormalities are seen in approximately one third of individuals; between 20% and 50% have seizures.36,37 Seven of 10 individuals ages 3–29 years undergoing polysomnography in the sleep laboratory had obstructive events.38 Dental abnormalities, including enamel defects, occur frequently. Excessive secretions are often noted.29 Individuals with Costello syndrome have very loose joints, particularly the fingers. Ulnar deviation of the wrists and fingers is common. Achilles tendons become very tight throughout childhood and often require surgical release.
Adolescents often show delayed or disordered puberty and may appear older than their chronologic age because of worsening kyphoscoliosis, sparse hair, and prematurely aged skin. In 17 adults ranging from age 16 to 40 years, all eight individuals who had a bone density measurement had abnormal results that suggested osteoporosis or osteopenia; three had bone pain, vertebral fractures, and height loss.39,40 Adult-onset gastroesophageal reflux was present in four individuals in the series of White et al.39; additional cases are known (personal observation). The reported adult height range is 135–150 cm.41
Tumor predisposition
Benign and malignant solid tumors occur with far greater frequency in individuals with Costello syndrome than in the general population. The overall tumor incidence is approximately 15% over the lifetime of individuals with an identified HRAS mutation.11 Rhabdomyosarcoma occurs most frequently, followed by neuroblastoma and transitional cell carcinoma of the bladder, and other solid tumors.4 Rhabdomyosarcoma and neuroblastoma are tumors of early childhood, presenting in Costello syndrome at ages comparable with the general population. In contrast, transitional cell carcinoma of the bladder occurs in older adults (70%, age >65 years) in the general population, whereas it occurs in adolescents with Costello syndrome. The ages at presentation in the three reported cases were 10, 11, and 16 years.
Prevalence and mortality
A formal epidemiologic study of Costello syndrome, with lifespan analysis, survival and cumulative mortality has not been performed. Lin et al.42 studied 146 mutation-confirmed patients, with at least another 85 clinically defined cases from the literature. The total is at least 220, and with additional unreported patients around the world, it is likely the figure approaches 300. The Costello Syndrome Family Network estimates that the birth prevalence in the United Kingdom is at least 1/500,000.7 As part of the recent cardiovascular study,42 deaths were reported in 10% and in the study and 20% of literature patients, providing preliminary descriptive data. Cardiorespiratory causes included HCM in all 11 (48%) accompanied by neoplasia, coronary artery fibromuscular dysplasia, and multifocal tachycardia in four. Neoplasia was noted at the cause of death in five (22%), pulmonary cause in two (9%), and multiorgan failure in five (four with HCM) (22%).
Genotype-phenotype correlations
Because few affected individuals with mutations other than p.Gly12Ser have been identified, limited genotype-phenotype correlations have been established. However, Kerr et al.12 suggested that the risk for malignant tumors may be higher in individuals with the p.Gly12Ala mutation (4/7 or 57%) than in those with the p.Gly12Ser variant (4/65 or 7%). Lo et al.43 suggested that a more severe neonatal phenotype may be associated with certain rare mutations, including p.Gly12Asp and p.Gly12Cys. In contrast, the possibility of a milder or attenuated phenotype was noted in individuals with p.Thr58Ile and p.Ala146Val.24 Distinctive phenotypic findings are associated with the p.Gly13Cys change and include dolichocilia (extremely long eye lashes, often requiring trimming) and loose anagen hair.8 Papillomata or multifocal atrial tachycardia have not yet been seen in individuals with p.Gly13Cys, and fewer have short stature. Compared with individuals with the most common p.Gly12Ser, these differences are statistically significant. In addition, no individual with p.Gly13Cys has developed a malignant tumor.8 The two individuals reported with p.Glu37dup share phenotypic findings including very sparse hair and facial features that appear less coarse than in most other Costello syndrome individuals, but a detailed clinical description was not provided.25 Of 146 patients with HRAS mutations whose cardiovascular status was reviewed, there was no apparent correlation between the specific mutation and the variables studied (HCM, multifocal tachycardia, and aortic dilation; Table 5 in Lin et al.42). In some cases, small numbers prevented formal statistical analysis.
One individual with somatic mosaicism (20–30% of DNA derived from buccal cells exhibited the HRAS mutation p.Gly12Ser, which was not detected in DNA derived from blood cells) had an atypical phenotype attributed to her mosaicism. Findings typical for Costello syndrome included intellectual disability, short stature, sparse hair, coarse facial features, nasal papillomata, and tight Achilles tendons. Atypical findings included microcephaly, streaky areas of skin hypo- and hyperpigmentation, and normal menarche with subsequent regular menses.44 Somatic mosaicism was seen in a father who showed a patchy distribution of skin findings suggestive of Costello syndrome and who transmitted the p.Gly12Ser change to his son with typical Costello syndrome.13 Mosaicism for the p.Gly12Ser mutation was noted in an individual with a typical presentation including apparent cutis laxa.14 As may be expected in an autosomal dominant trait, somatic mosaicism can thus be associated with a range of findings including patchy or segmental distribution, typical phenotypic presentation, and transmission to offspring and recurrence in siblings.44
Treatment of manifestations
Medical care for individuals with Costello syndrome aims to improve overall health and development through early identification of anticipated medical problems and prompt treatment. Its ultimate goal is to improve quality of life for affected individuals and their families.45
Neonatal hypoglycemia has frequently been reported, and a high level of suspicion should be maintained. Rarely, hypoglycemia occurs in older individuals and may present with seizures. Under these circumstances, GH deficiency needs to be excluded as the underlying cause.46 Hypoglycemic episodes unresponsive to GH therapy responded well to cortisone replacement in another individual.47 Thus, cortisol deficiency may also be considered. Because of the severe feeding difficulties, most infants require nasogastric or gastrostomy feeding. Because of gastroesophageal reflux and irritability, Nissen fundoplication is often performed. Pyloric stenosis is treated surgically. Severe irritability associated with feeding and an inability to sleep has anecdotally improved by use of benzodiazepines, resulting in decreased irritability and weight gain. Anecdotally, affected children have very high caloric needs. Even after nutrition is improved through supplemental feeding, growth retardation persists. GH deficiency can often be documented on stimulation testing and warrants replacement therapy. Although linear growth may benefit from GH replacement, the main objective of replacement therapy is to prevent secondary complications, such as hypoglycemia and low muscle mass. Orthopedic issues can include congenital hip dysplasia, scoliosis, and kyphoscoliosis. Joint contractures, often affecting elbows, wrists, and fingers are treated through physical and occupational therapy or by early bracing. Surgical tendon lengthening, usually of the Achilles tendon, is often required in late childhood. Low bone density is common in young adults with Costello syndrome; and bone density assessment is recommended as a baseline, with follow-up depending on the initial result. Osteoporosis may be ameliorated by physical activity and increased vitamin D and calcium intake, or require treatment with pamidronate.
The high risk for postnatal development of a Chiari I malformation and its sequelae warrant a brain imaging study, preferably a magnetic resonance imaging, at the time of diagnosis and a repeat scan 1–2 years later or whenever the patient becomes symptomatic.28 In older individuals, headaches, changes in gait, and other neurologic symptoms may prompt imaging of the central nervous system and spinal cord. A symptomatic Chiari I malformation will require surgical intervention, typically a posterior fossa decompression. Tethered cord is common in individuals with Costello syndrome and may present with loss of bowel and bladder function or gait disturbance, it is treated surgically. Developmental disability should be addressed by early-intervention programs and individualized learning strategies. Speech delay and expressive language limitations will benefit from appropriate therapy and an appropriate educational plan. As for every individual with developmental delay, an eye and hearing evaluation is indicated, and medical problems are treated as necessary. Alternate means of communication should be considered if expressive language is significantly limited. Anxiety occurs more frequently than in the general population, and it may benefit from supportive therapies, appropriate educational expectations, and use of standard medications. The identification of individual cognitive and adaptive strengths, and the development of an educational plan building on these strengths, may improve independent living skills and will result in improved quality of life.
We continue to recommend that all individuals with Costello syndrome, especially those with an identified cardiovascular abnormality, should be followed up by a cardiologist who is aware of the spectrum of cardiac disease and its natural history.30,42 Rather than propose a unique set of guidelines, we advise providers and families to receive individualized care by a pediatric cardiologist, transitioning to an adult specialist following the “best practices” for the particular defect. General guidelines (Table 4 in Lin et al.42) can be pragmatically dichotomized based on the presence or absence of HCM, with close evaluation in the first 2 years of life depending on the severity of hypertrophy, subsequent annual examinations, and appropriate risk stratification. We emphasized in that article and remind providers that it is beyond the scope of a genetics article to review the complex decision making for treating HCM or atrial tachycardia. The benefit of recent research42 is that it adds modest natural history data, although ongoing studies of older individuals will be necessary to monitor for late-onset HCM progression or arrhythmia.42 The treatment of tachycardia in Costello syndrome has not been formally studied, and anecdotal reports may not be broadly applied. Malignant nonreentrant tachycardia may require electrophysiologic catheterization, aggressive antiarrhythmic drugs, and ablation. Pharmacologic and surgical treatment (myectomy) has been used to treat severe cardiac hypertrophy. Individuals with Costello syndrome and severe cardiac problems may choose to wear a Medic Alert® bracelet.
The benign papillomata usually appear in the perinasal region and less commonly in the perianal region, torso, and extremities. Although they are mostly of cosmetic concern, papillomata may give rise to irritation or inflammation in hard-to-clean body regions and may be removed, as appropriate. Recurrent facial papillomata have been successfully managed with regular dry ice removal. Malignant tumors, most commonly embryonal rhabdomyosarcoma, occur with increased frequency in Costello syndrome, and this led to the proposal of a screening protocol. Abdominal and pelvic ultrasound and urine testing for catecholamine metabolites and hematuria were suggested.48 However, a subsequent report49 on elevated catecholamine metabolites in individuals with Costello syndrome without an identifiable tumor concluded that screening for abnormal catecholamine metabolites is not helpful. Serial abdominal and pelvic ultrasound screening for rhabdomyosarcoma and neuroblastoma was proposed every 3–6 months until age 8–10 years. Urinalysis for hematuria was suggested annually beginning at age 10 years to screen for bladder cancer.48 Neither of these screening approaches has yet been proven to be beneficial; however, studies are ongoing. The most important factor for early tumor detection remains parental and physician awareness of the increased cancer risk. Treatment of malignant tumors follows standard protocols.
The rasopathies clinic: a pragmatic care model
Costello syndrome affects multiple organ systems and requires medical care expertise from numerous specialty areas, including cardiology, gastroenterology, neurology and neurosurgery, orthopedics, urology, developmental medicine and therapeutic services, and often oncology. With approximately 300 reported patients, Costello syndrome remains rare, and most specialists will see very few affected individuals throughout their career and will thus have little personal experience with this condition. However, the medical issues seen in Costello syndrome may resemble those seen in other rasopathies, for example, the HCM in Noonan syndrome and CFC syndrome, the skin findings shared with CFC syndrome, or the low bone density and increased tumor risk in NF1. In fact, the phenotype of Costello, Noonan, and CFC syndromes shows such significant overlap during infancy that a clear distinction cannot always be made on clinical criteria alone, but rather molecular testing is necessary. This overlap in the physical manifestations is easily understood based on the close relationship of these conditions through their shared mechanism of Ras/MAPK pathway dysregulation. In light of the many similar medical issues for individuals with any rasopathy, a clinic model combining patients with these conditions is beneficial as it allows the specialists to become more familiar with these conditions.50 Two of the rasopathies, NF1 and Noonan syndrome, are relatively common, and most healthcare institutions are familiar with their phenotype and medical issues. Specialists, such as cardiologist caring for Noonan syndrome individuals, can become familiar with the phenotypic presentation and may be more comfortable caring for a patient with Costello syndrome. To a large degree, this occurs informally, but the designation of a formal clinic will focus attention, resources, and patient care. Additional benefits of a combined rasopathy clinic include the formation of a local rasopathy advocacy group, which could complement the national (Costello Syndrome Family Network) and international (Costellokids.org) family support organizations. Furthermore, such clinics may provide an organizational structure for future treatment trials.51 Discussion about research has already occurred at three International Meetings under the name “Genetic Syndromes of the Ras/MAPK Pathway.”
Disclosure
The authors declare no conflict of interest.
References
Costello JM . A new syndrome. NZ Med J 1971;74:397.
Costello JM . A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J 1977;13:114–118.
Gripp KW, Lin AE . Costello syndrome. In: GeneReviews at GeneTests: Medical Genetics Information Resource [database online]. University of Washington: Seattle, 1997–2009. http://www.genetests.org/. Accessed 1 May 2011.
Gripp KW . Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005;137C:72–77.
Aoki Y, Niihori T, Kawame H,et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 2005;37:1038–1040.
Rasopathy Network, 2011. http://ras-pathway-syndromes.com/. Accessed 26 April 2011.
Costello Kids Support Group. http://www.costellokids.com/. Accessed 5 June 2011.
Gripp KW, Hopkins E, Sol-Church K,et al. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med Genet A 2011;155A:706–716.
Kerr B, Allanson J, Delrue MA,et al. The diagnosis of Costello syndrome: nomenclature in Ras/MAPK pathway disorders. Am J Med Genet A 2008;146A:1218–1220.
Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA . HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 2006;140:8–16.
Gripp KW, Lin AE, Stabley DL,et al. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A 2006;140:1–7.
Kerr B, Delrue MA, Sigaudy S,et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet 2006;43:401–405.
Sol-Church K, Stabley DL, Demmer LA,et al. Male-to-male transmission of Costello syndrome: G12S HRAS germline mutation inherited from a father with somatic mosaicism. Am J Med Genet A 2009;149A: 315–321.
Girisha KM, Lewis LE, Phadke SR, Kutsche K . Costello syndrome with severe cutis laxa and mosaic HRAS G12S mutation. Am J Med Genet A 2010;152A:2861–2864.
Rauen KA . Distinguishing Costello versus cardio-facio-cutaneous syndrome: BRAF mutations in patients with a Costello phenotype. Am J Med Genet A 2006;140:1681–1683.
Gripp KW, Lin AE, Nicholson L,et al. Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome from Costello syndrome. Am J Med Genet A 2007;143A:1472–1480.
Quezada E, Gripp KW . Costello syndrome and related disorders. Curr Opin Pediatr 2007;19:636–644.
Kerr B, Mucchielli ML, Sigaudy S,et al. Is the locus for Costello syndrome on 11p? J Med Genet 2003;40:469–471.
Rosenberger G, Meien S, Kutsche K . Oncogenic HRAS mutations cause prolonged PI3K signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutat 2009;30: 352–362.
Sol-Church K, Gripp KW . The molecular basis of Costello syndrome. In: Zenker M (ed). Noonan Syndrome and Related Disorders: A Matter of Deregulated Ras Signaling (monographs in human genetics). S. Karger: Switzerland, 2009:94–103.
Lin AE, Rauen KA, Gripp KW, Carey JC . Clarification of previously reported Costello syndrome patients. Am J Med Genet A 2008;146:940–943.
van der Burgt I, Kupsky W, Stassou S,et al. Myopathy caused by HRAS germline mutations: implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J Med Genet 2007;44: 459–462.
Zampino G, Pantaleoni F, Carta C,et al. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat 2007;28:265–272.
Gripp KW, Innes AM, Axelrad ME,et al. Costello syndrome associated with novel germline HRAS mutations: an attenuated phenotype? Am J Med Genet A 2008;146A:683–690.
Gremer L, De Luca A, Merbitz-Zahradnik T,et al. Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet 2010;19:790–802.
Lin AE, O’Brien B, Demmer LA,et al. Prenatal features of Costello syndrome: ultrasonographic findings and atrial tachycardia. Prenat Diagn 2009;29:682–690.
Smith LP, Podraza J, Proud VK . Polyhydramnios, fetal overgrowth, and macrocephaly: prenatal ultrasound findings of Costello syndrome. Am J Med Genet A 2009;149A:779–784.
Gripp KW, Hopkins E, Doyle D, Dobyns WB . High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A 2010;152A:1161–1168.
Johnson JP, Golabi M, Norton ME,et al. Costello syndrome: phenotype, natural history, differential diagnosis, and possible cause. J Pediatr 1998;133:441–448.
Lin AE, Grossfeld PD, Hamilton RM,et al. Further delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet 2002;111:115–129.
Tidyman WE, Lee HS, Rauen KA . Skeletal muscle pathology in Costello and cardio-facio-cutaneous syndromes: developmental consequences of germline Ras/MAPK activation on myogenesis. Am J Med Genet C Semin Med Genet 2011;157:104–114.
Axelrad ME, Glidden R, Nicholson L, Gripp KW . Adaptive skills, cognitive, and behavioral characteristics of Costello syndrome. Am J Med Genet A 2004;128A:396–400.
Axelrad ME, Nicholson L, Stabley DL, Sol-Church K, Gripp KW . Longitudinal assessment of cognitive characteristics in Costello syndrome. Am J Med Genet A 2007;143A:3185–3193.
Axelrad ME, Schwartz DD, Fehlis JE,et al. Longitudinal course of cognitive, adaptive, and behavioral characteristics in Costello syndrome. Am J Med Genet A 2009;149A:2666–2672.
Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW . Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet 2011;157:115–122.
Delrue MA, Chateil JF, Arveiler B, Lacombe D . Costello syndrome and neurological abnormalities. Am J Med Genet A 2003;123A:301–305.
Kawame H, Matsui M, Kurosawa K,et al. Further delineation of the behavioral and neurologic features in Costello syndrome. Am J Med Genet A 2003;118A:8–14.
Della Marca G, Vasta I, Scarano E,et al. Obstructive sleep apnea in Costello syndrome. Am J Med Genet A 2006;140:257–262.
White SM, Graham JM Jr, Kerr B,et al. The adult phenotype in Costello syndrome. Am J Med Genet A 2005;136:128–135.
Stevenson DA, Yang FC . The musculoskeletal phenotype of the RASopathies. Am J Med Genet C Semin Med Genet 2011;157:90–103.
Hennekam RC . Costello syndrome: an overview. Am J Med Genet C Semin Med Genet 2003;117C:42–48.
Lin AE, Alexander ME, Colan SD,et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet A 2011;155A:486–507.
Lo IF, Brewer C, Shannon N,et al. Severe neonatal manifestations of Costello syndrome. J Med Genet 2008;45:167–171.
Gripp KW, Stabley DL, Nicholson L, Hoffman JD, Sol-Church K . Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A 2006;140:2163–2169.
Hopkins E, Lin AE, Krepkovich KE,et al. Living with Costello syndrome: quality of life issues in older individuals. Am J Med Genet A 2010;152A: 84–90.
Gripp KW, Scott CI Jr, Nicholson L, Figueroa TE . Second case of bladder carcinoma in a patient with Costello syndrome. Am J Med Genet 2000;90:256–259.
Gregersen N, Viljoen D . Costello syndrome with growth hormone deficiency and hypoglycemia: a new report and review of the endocrine associations. Am J Med Genet A 2004;129A:171–175.
Gripp KW, Scott CI Jr, Nicholson L,et al. Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. Am J Med Genet 2002;108:80–87.
Gripp KW, Kawame H, Viskochil DH, Nicholson L . Elevated catecholamine metabolites in patients with Costello syndrome. Am J Med Genet A 2004;128A:48–51.
Rauen KA, Schoyer L, McCormick F,et al. Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am J Med Genet A 2010;152A:4–24.
Rauen KA, Banerjee A, Bishop WR,et al. Costello and cardio-faciocutaneous syndromes: moving toward clinical trials in RASopathies. Am J Med Genet Part C Semin Med Genet 2011;157:136–146.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gripp, K., Lin, A. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med 14, 285–292 (2012). https://doi.org/10.1038/gim.0b013e31822dd91f
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.0b013e31822dd91f
Keywords
This article is cited by
-
A systematic review, meta-analysis, and network analysis of diagnostic microRNAs in glaucoma
European Journal of Medical Research (2023)
-
Fatal leukodystrophy in Costello syndrome: a case report
BMC Pediatrics (2023)
-
Clinical profiling of MRD48 and functional characterization of two novel pathogenic RAC1 variants
European Journal of Human Genetics (2023)
-
Social behavior in RASopathies and idiopathic autism
Journal of Neurodevelopmental Disorders (2022)
-
Structure–function analysis of the SHOC2–MRAS–PP1C holophosphatase complex
Nature (2022)
