
Abstract
Purpose
To assess the safety and efficacy of an eye drop combining osmoprotectants, carboxymethylcellulose and hyaluronic acid (O/CMC/HA) in reducing symptomatic, moderate to severe dry eye, compared with HA.
Methods
In this investigator-masked, randomised study, patients instilled 1–2 drops/eye of O/CMC/HA or HA (2–6 times/day) for 3 months. Primary endpoint: mean change in Global Ocular Staining Score (GOSS) from baseline at day 35. Noninferiority of O/CMC/HA was tested in the per-protocol population; if achieved, superiority was tested in the intent-to-treat population. Secondary efficacy endpoints: mean change from baseline in GOSS, Ocular Surface Disease Index (OSDI), Schirmer score, tear break-up time (TBUT), corneal/conjunctival staining, conjunctival hyperaemia, symptoms, and patient/investigator assessments.
Results
Baseline characteristics were comparable between groups (n=40 each). O/CMC/HA was noninferior (and not superior) to HA based on similar GOSS reductions from baseline at day 35 and month 3 in both groups (P=0.778, day 35, per-protocol population). Overall, O/CMC/HA and HA provided similar reductions in OSDI, Schirmer score, TBUT, corneal staining and hyperaemia from baseline at 35 days (P≥0.155). More patients reported less severe stinging/burning, sandiness/grittiness, and painful/sore eyes at month 3 with O/CMC/HA (P≤0.039), and more rated the dropper bottle easy to use (87.5%), compared with HA (46.2%; P=0.002). Other patient and investigator assessments were similar between groups. O/CMC/HA and HA were well tolerated.
Conclusions
O/CMC/HA is noninferior to HA in improving objective signs of dry eye, with potential advantages for subjective symptoms and patient acceptance.
Similar content being viewed by others

Introduction
Dry eye is a multifactorial, often chronic disease of the tears and ocular surface that affects up to 30% of the global population aged ≥50 years.1 It is accompanied by hyperosmolarity of the tear film (due to reduced tear flow and/or increased evaporation) and inflammation of the ocular surface that can damage the ocular surface if left untreated.2, 3 Dry eye also leads to impairments in vision-related quality of life, including difficulties reading, working, using a computer, watching television, and driving.4 The burden of dry eye not only includes direct costs associated with healthcare resource utilisation, but also indirect costs related to lost work time/productivity.1, 5
Used alone or combined with other treatments, artificial tears are standard therapy at all stages of dry eye.6 Water-retentive polymers such as carboxymethylcellulose (CMC) and hyaluronic acid (HA) have both been used in artificial tear formulations to protect the ocular surface7, 8 by maintaining/restoring stability of the tear film.9 In addition, HA has been shown to modulate the inflammatory response.10 More recently, osmoprotectants erythritol, L-carnitine and glycerine have been added to CMC11 to reduce the cellular stress level of the ocular surface,12, 13 but to date, these ingredients have not been combined with HA.
An artificial tear solution containing CMC 0.5%, HA 0.1%, and osmoprotectants (glycerine and erythritol3, 12, 14) has been introduced for the treatment of dry eye disease (O/CMC/HA; Optive Fusion; Allergan plc, Dublin, Ireland). This combination has potential synergistic effects resulting in improved viscosity and hydration, as well as enhanced ocular surface integrity, compared with CMC or HA alone.15 The present study evaluated the efficacy and safety of this multi-ingredient, preserved artificial tear formulation in reducing the signs and symptoms of moderate to severe dry eye, compared with a preservative-free, hypotonic formulation of HA 0.18% (Vismed Multi; TRB Chemedica International S.A., Geneva, Switzerland). The clinical efficacy hypothesis was that in patients with symptomatic dry eye, the effect of O/CMC/HA (administered 2–6 times daily as needed) on ocular surface integrity was not inferior to that of HA, as measured by a mean decrease in global ocular staining score (GOSS) from baseline.
Materials and methods
Study design
This was a multicentre, investigator-masked, randomised, 3-month, noninferiority study (ClinicalTrials.gov identifier: NCT02117687). The protocol was approved by local investigational review boards or independent ethics committees before study start, and all patients provided written informed consent before initiating treatment. The study was conducted between May 2014 and March 2015 in compliance with the International Conference on Harmonisation guidelines for Good Clinical Practice and the Declaration of Helsinki, and involved 14 centres in France and one centre in the United Kingdom.
Study visits were scheduled at day 1 (visit 1/baseline), day 35+7 days (visit 2), and month 3±7 days (visit 3). Patients were also contacted by telephone on day 8+2 days.
Participants
Eligible patients were ≥18 years old and had moderate to severe symptomatic dry eye (defined according to the International Dry Eye Workshop classification2) with a baseline Ocular Surface Disease Index (OSDI) score ≥22 on a 0–100 scale.16 They had used artificial tears in both eyes for ≥3 months before inclusion, had been using preservative-free artificial tears at least twice daily for ≥2 weeks at baseline, and had at least one eye with both of the following conditions at baseline: GOSS ≥4 and ≤9 on the 0–15 Oxford scale,17, 18 and a Schirmer score (without anaesthesia) ≥3 and ≤9 mm/5 min or three consecutive tear break-up time (TBUT) tests ≤10 s.
Key ophthalmic exclusion criteria were best-corrected visual acuity (BCVA) <20/200; moderate to severe blepharitis; severe dry eye with eyelid abnormalities, corneal disorder/abnormality that affects cornea sensitivity (or complete coverage of the tear film), ocular surface metaplasia, filamentous keratitis, or corneal vascularisation; history/active signs of ocular trauma, infection or inflammation (within 3 months of visit 1), or severe/serious ocular conditions that could prevent study completion; active signs of ocular allergic disease or ocular herpes (within 2 years of visit 1); surgery involving a limbal or corneal incision within 12 months of visit 1; use of intra- or periocular medications or punctal plugs (or lacrimal punctum cauterisation); and anticipated use of any ophthalmic product, except the study product, between visit 1 and visit 3.
Treatment and assessments
At the baseline visit, patients were randomised (1:1) to O/CMC/HA or HA treatment, based on a randomisation scheme provided to each site. Patients were instructed to stop using their preservative-free artificial tears and start administering 1–2 drops of study medication in each eye 2–6 times each day (as needed) for 3 months. They were also instructed not to instil the study product within the hour preceding scheduled assessment visits, and not to reveal the nature of their study product to the investigator.
GOSS grading was based on the severity of corneal fluorescein staining, as well as nasal conjunctiva and temporal conjunctiva lissamine green staining, each graded from 0 to 5 according to the Oxford staining chart.17, 18, 19 The OSDI evaluated the frequency of dry eye symptoms in the week preceding each study visit; scores between 23 and 32 indicate moderate dry eye, and scores >32 indicate severe dry eye.16 Conjunctival hyperaemia was evaluated using slit-lamp biomicroscopy (without pupil dilation), and graded on a five-point photographic scale (0=none, 0.5=trace, 1=mild, 2=moderate, 3=severe). Tear production was assessed using the Schirmer test (without anaesthesia) and TBUT. The same evaluator was to perform the evaluations throughout the study.
Bilateral assessment of dry eye symptoms by patients was based on a questionnaire using a five-point Likert Scale ranging from 0 (no discomfort) to 4 (very severe). Patients also rated treatment acceptability on a five-point scale (strongly agree, agree, neither agree nor disagree, disagree, or strongly disagree), as well as their work productivity and activity impairment, and recorded how many times per day they used their study treatment to relieve their dry eye symptoms during the week preceding a study visit.
All examination procedures and assessments were performed on the scheduled visit days and in the following order: patient assessment of treatment acceptability, OSDI, patient assessment of symptoms, work productivity and activity impairment, BCVA, TBUT, corneal staining, biomicroscopy with hyperaemia grading, conjunctival staining, Schirmer test, and investigator global evaluation of treatment efficacy and safety.
Outcome measures and analyses
The primary efficacy endpoint was the mean change in GOSS from baseline at day 35 in the study eye. The study eye was the eye with greater GOSS at baseline, or the right eye if both eyes had equal GOSS. Noninferiority of O/CMC/HA (compared with HA) was tested in the per-protocol (PP) population (ie, all randomised patients who received ≥1 dose of study treatment, had ≥1 follow-up visit, and no major protocol violations). A two-sided 95% confidence interval (CI) of the treatment difference at day 35 was determined, based on a two-way analysis of covariance (ANCOVA) model with change from baseline as main effect, and site and baseline GOSS as covariates. If the upper limit was less than or equal to the prespecified two-grade margin, O/CMC/HA was considered noninferior to HA, and a superiority test followed (with a two-sided significance level of 0.05) in the intent-to-treat (ITT) population (ie, all randomised patients who received ≥1 dose of study treatment and had ≥1 follow-up visit).
Secondary efficacy endpoints included mean change from baseline in GOSS at month 3, mean change in OSDI, corneal staining, conjunctival hyperaemia, dry eye symptoms, work productivity/activity impairment and investigator global efficacy assessment, as well as daily use, at day 35 and month 3. Changes in TBUT and Schirmer test score from baseline were assessed at day 35, along with treatment acceptability. Secondary efficacy data from the ITT and PP populations were summarised by treatment and study/fellow eye at each timepoint. For appropriate secondary endpoints, the comparison of treatment groups was performed using a two-way ANCOVA including factors for treatment, baseline value (where appropriate) and site for continuous variables, or the Cochran–Mantel–Haenszel test with stratification by site for categorical variables.
Safety data were summarised using descriptive statistics for all patients in the safety population (ie, all randomised patients who received ≥1 dose of study treatment). Safety measures included adverse events (AEs), BCVA, biomicroscopy, and investigator global assessment of treatment safety.
A sample size of 40 patients per treatment group (80 total) was planned to provide 90% power to determine noninferiority based on the mean change in GOSS at day 35, using a one-sided, two-sample t test with an alpha of 0.025 and an estimated common standard deviation (SD) of 2.5. The study was not powered for secondary endpoints. Data and associated protocols are available upon request to Allergan plc.
Results
Patient disposition and demographics
The ITT population consisted of 80 patients randomised to O/CMC/HA (n=40) or HA (n=40). Among those, 97.5 and 90.0% completed treatment with O/CMC/HA and HA, respectively (Supplementary Table 1); no AEs led to study discontinuations of O/CMC/HA, whereas moderate conjunctivitis (n=1, not treatment-related) and mild eye irritation (n=1, treatment-related) resulted in early discontinuation of HA. Overall, at baseline, 13.8% of patients had Sjögren’s syndrome, 83.8% had a GOSS ≤6, and 36.3% had Schirmer test score of ≤5 mm/5 min. There were no statistically significant baseline differences in gender, age, Sjögren’s syndrome status, GOSS, or Schirmer test scores between treatment groups (Table 1).
The PP population consisted of 66 patients (O/CMC/HA, n=35; HA, n=31; Supplementary Table 1). Baseline demographics and characteristics were similar to those of the ITT population, and not statistically different between groups.
Efficacy
In the PP population, the mean change in GOSS from baseline at day 35 (primary endpoint) demonstrated that O/CMC/HA was noninferior to HA (Table 2); the upper limit of the 95% CI for the between-group difference was within the prespecified two-grade margin of noninferiority. Mean baseline GOSS was similar between the O/CMC/HA (5.2±1.1) and HA (5.2±1.2) groups, and a reduction was observed in both groups over the course of the study to 2.7±2.2 and 2.8±2.3 at 3 months, respectively. Both treatment groups achieved significant reductions in GOSS from baseline at day 35 and month 3 (Table 2).
These results were confirmed in the ITT population. Superiority of O/CMC/HA over HA was not demonstrated based on the mean change in GOSS from baseline at day 35 in this population (Table 2). Mean baseline GOSS was similar in the O/CMC/HA (5.4±1.3) and HA (5.4±1.6) groups, and decreased to 3.2±2.7 and 3.1±2.5 at month 3, respectively. Significant reductions in GOSS from baseline were observed at day 35 and month 3 in both groups (Table 2).
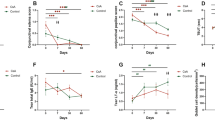
In the ITT population, the mean (SD) baseline OSDI scores were 46.1 (22.8) in the O/CMC/HA group and 47.6 (17.3) in the HA group, and decreased at day 35 and month 3. There was, however, no statistically significant difference in change from baseline between groups (Figure 1a). Results were similar in the PP population (P≥0.270).

Change from baseline in OSDI (a) and corneal staining (b) in the intent-to-treat population. HA, hyaluronic acid; O/CMC/HA, osmoprotectants/carboxymethylcellulose/hyaluronic acid; OSDI, ocular surface disease index.
Mean (SD) baseline corneal staining was 1.7 (0.7) in the O/CMC/HA group and 1.9 (0.8) in the HA group. Corneal staining decreased in both groups at day 35 and month 3, but the between-group differences did not reach statistical significance (Figure 1b). Results were similar for the PP population (P≥0.222).
Changes in the Schirmer scores and TBUT from baseline at day 35 were small and not clinically meaningful. In the ITT population, mean baseline Schirmer score in the study eye was 10.7±9.7 mm/5 min in the O/CMC/HA group versus 9.2±7.5 mm/5 min in the HA group. Changes from baseline scores were 0.0±7.3 and 1.2±6.1 at day 35, respectively, with a between-treatment difference of 0.3 (95% CI: −2.5, 3.0; P≥0.850). Mean baseline TBUT in the study eye was 5.4±2.6 s in the O/CMC/HA group and 5.4±2.9 s in the HA group. Changes from baseline were 0.6±1.7 and 0.6±1.9, respectively, with a between-treatment difference of 0.0 (95% CI: −0.6, 0.6; P=0.991). Results were similar in the PP population (P=0.584 for Schirmer scores and P=0.978 for TBUT).
Conjunctival hyperaemia improved over time in both groups, and more patients had no hyperaemia at month 3 in the O/CMC/HA group, but the differences did not reach statistical significance at day 35 or month 3 in either the ITT or PP populations (Supplementary Table 2). Mean daily use of the eye drops was similar for O/CMC/HA and HA (mean: 3.8±1.2 and 3.6±1.0, respectively) and remained stable throughout the study.
Safety and tolerability
Both eye drops were well tolerated, and no unexpected AEs were noted. A total of 36 AEs were reported by 21 patients (26.3%) and 16 treatment-related AEs (all of which were mild or moderate in severity) were reported by 9 patients (11.3%). The most common ocular AEs were eye pruritus in the O/CMC/HA group (n=2, 5.0%), and dry eye (n=2, 5.0%) and eye irritation (n=2, 5.0%) in the HA group (Table 3). The mean change (SD) in BCVA from baseline at month 3 on a Snellen chart was −0.03 (1.19) for O/CMC/HA and 0.13 (0.47) for HA, and there were no unexpected findings on biomicroscopy. Study investigators rated the safety of both eye drops as satisfactory or very satisfactory in >94% of patients at both timepoints. There were no statistically significant differences in ratings between groups at day 35 (P=0.801) and month 3 (P=0.557).
Patient questionnaires and assessments
The dry eye symptom questionnaires indicated that stinging/burning, itching, sandiness/grittiness, blurred vision, dryness, light sensitivity, and painful/sore eyes improved over time in both groups. More patients reported less severe stinging/burning, sandiness/grittiness, and painful/sore eyes at month 3 with O/CMC/HA than with HA (P≤0.039 for each symptom) (Figure 2a). Results were also statistically significant for painful/sore eyes at day 35 (P=0.035) and for light sensitivity at month 3 (P=0.027).

Patient assessment of dry eye symptoms (a) and treatment acceptability (b). HA, hyaluronic acid; ITT, intent-to-treat; O/CMC/HA, osmoprotectants/carboxymethylcellulose/hyaluronic acid.
More patients agreed or strongly agreed that the O/CMC/HA dropper bottle (87.5%) was easy to use, compared with the HA bottle (46.2%; P=0.002), whereas similar proportions of patients in both groups agreed or strongly agreed that they liked using their eye drops (P=0.491), and that their eyes felt comfortable after using them (P=0.993; Figure 2b).
The work productivity and activity impairment questionnaire assessed the impact of dry eye on the ability to work and perform regular activities over the preceding 7 days. Some questionnaire items, however, were not applicable to the entire population because not all patients were employed full time. There was a trend toward improvement of absenteeism, presenteeism and productivity over the course of the study; the between-group difference closest to the significance threshold was for productivity at day 84 (P=0.053).
Discussion
The results of this randomised, investigator-masked study demonstrate that in patients with moderate to severe dry eye, the combination of erythritol, CMC 0.5%, HA 0.1%, and glycerine in a preserved artificial tear formulation is noninferior to nonpreserved HA 0.18% alone in improving objective signs of dry eye as measured by the change in GOSS from baseline at day 35 in the PP population (primary endpoint). Both artificial tear formulations reduced GOSS over the course of the study, but between-group differences in change from baseline at day 35 did not reach statistical significance (P=0.778). Analysis of both the PP and ITT populations is required to draw valid conclusions from a noninferiority analysis,20 and similar reductions in GOSS from baseline at day 35 occurred in the ITT population.
Our primary assessment timepoint of 35 days was chosen as 4–5 weeks of treatment is a commonly recognised period to assess tear substitutes owing to their short duration of action on the ocular surface. Moreover, this time period should limit the impact of fluctuations associated with the natural course of dry eye.
Because noninferiority was demonstrated for the primary endpoint, a superiority analysis was also conducted on the GOSS change from baseline at day 35, but superiority was not achieved. Both artificial tear formulations demonstrated objective improvement of other efficacy endpoints throughout the course of the study. There were no statistically significant differences between treatments for the mean change from baseline in OSDI, corneal fluorescein staining, or conjunctival hyperaemia. Neither formulation demonstrated clinically meaningful improvements in mean Schirmer scores or TBUT, and both formulations were well tolerated, demonstrating similar AE profiles. Most investigators rated the safety of the artificial tear formulations as satisfactory or very satisfactory.
In addition to the objective efficacy measures evaluated in this study, patients completed questionnaires that subjectively measured changes in dry eye symptoms and acceptability of the two artificial tear formulations. Perceived efficacy and treatment acceptability are important metrics, particularly in chronic conditions, such as dry eye, that require long-term therapy. Both O/CMC/HA and HA improved symptoms of stinging/burning, itching, sandiness/grittiness, blurred vision, dryness, light sensitivity, and painful/sore eyes over the course of the 3-month study. Compared with HA, a greater percentage of patients reported significantly less severe stinging/burning, sandiness/grittiness, and painful/sore eyes at month 3 with the O/CMC/HA formulation. In addition, significantly fewer patients experienced painful/sore eyes at day 35 in the O/CMC/HA group, compared with the HA group. More patients reported that the O/CMC/HA eye drop bottle was easy to use, compared with the HA dropper bottle, which is notable considering a recent glaucoma study that showed statistically significant variability in the force required to dispense drops from 21 different bottle designs.21 In contrast, the proportion of patients who liked using the eye drops or whose eyes felt comfortable after using the eye drops was similar for both groups.
Earlier studies have compared the efficacy and safety of eye drops containing CMC only to HA alone9, 11, 22, 23, 24 or combinations of CMC, HA and osmoprotectants.25 Three studies comparing CMC-based eye drops with formulations of HA alone demonstrated similar efficacy and safety.11, 22, 24 Other studies showed that CMC eye drops resulted in a significantly greater reduction from baseline in conjunctival staining, compared with HA.9, 23 When CMC alone was compared with O/CMC/HA, no statistically significant differences in mean change from baseline OSDI scores were noted in the overall population. However, when patients were stratified by baseline disease severity, O/CMC/HA was more effective than CMC alone in patients with more severe disease (combined corneal/conjunctival staining scores ≥14).25 In the present study, when HA alone was compared with O/CMC/HA, no significant differences were found in the reduction of ocular staining from baseline, but there were advantages in favour of O/CMC/HA concerning several types of subjective symptoms.
Overall, our findings showed that O/CMC/HA is noninferior to HA alone in improving objective signs of dry eye disease, safe, and well tolerated. The O/CMC/HA combination also improved some subjective signs of dry eye disease (ie, stinging/burning, sandiness/grittiness, and painful/sore eyes) at month 3 and more patients found the O/CMC/HA dropper bottle easy to use, compared with the HA dropper bottle. However, studies are needed in specific high-risk patient populations, including patients with more severe dry eye disease, to fully define the place of O/CMC/HA in the armamentarium of therapies for dry eye. Future studies should also identify optimal dosing frequency of O/CMC/HA and compare its efficacy and safety with other HA-containing formulations.

References
The epidemiology of dry eye disease: report of the Epidemiology Subcommittee of the International Dry Eye WorkShop. 2007 Ocul Surf 2007; 5: 93–107.
The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop. Ocul Surf 2007; 5: 75–92.
Baudouin C, Aragona P, Messmer EM, Tomlinson A, Calonge M, Boboridis KG et al. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: proceedings of the OCEAN group meeting. Ocul Surf 2013; 11: 246–258.
Miljanovic B, Dana R, Sullivan DA, Schaumberg DA . Impact of dry eye syndrome on vision-related quality of life. Am J Ophthalmol 2007; 143: 409–415.
Nichols KK, Bacharach J, Holland E, Kislan T, Shettle L, Lunacsek O et al. Impact of dry eye disease on work productivity, and patients' satisfaction with over-the-counter dry eye treatments. Invest Ophthalmol Vis Sci 2016; 57: 2975–2982.
Lemp MA . Management of dry eye disease. Am J Manag Care 2008; 14 (3 Suppl): S88–101.
Yokoi N, Komuro A, Nishida K, Kinoshita S . Effectiveness of hyaluronan on corneal epithelial barrier function in dry eye. Br J Ophthalmol 1997; 81: 533–536.
Simmons PA, Vehige JG . Clinical performance of a mid-viscosity artificial tear for dry eye treatment. Cornea 2007; 26: 294–302.
Guillon M, Maissa C, Ho S . Evaluation of the effects on conjunctival tissues of Optive eyedrops over one month usage. Cont Lens Anterior Eye 2010; 33: 93–99.
Oh HJ, Li Z, Park SH, Yoon KC . Effect of hypotonic 0.18% sodium hyaluronate eyedrops on inflammation of the ocular surface in experimental dry eye. J Ocul Pharmacol Ther 2014; 30: 533–542.
Baudouin C, Cochener B, Pisella PJ, Girard B, Pouliquen P, Cooper H et al. Randomized, phase III study comparing osmoprotective carboxymethylcellulose with sodium hyaluronate in dry eye disease. Eur J Ophthalmol 2012; 22: 751–761.
Corrales RM, Luo L, Chang EY, Pflugfelder SC . Effects of osmoprotectants on hyperosmolar stress in cultured human corneal epithelial cells. Cornea 2008; 27: 574–579.
Hua X, Su Z, Deng R, Lin J, Li DQ, Pflugfelder SC . Effects of L-carnitine, erythritol and betaine on pro-inflammatory markers in primary human corneal epithelial cells exposed to hyperosmotic stress. Curr Eye Res 2015; 40: 657–667.
Monaco G, Cacioppo V, Consonni D, Troiano P . Effects of osmoprotection on symptoms, ocular surface damage, and tear film modifications caused by glaucoma therapy. Eur J Ophthalmol 2011; 21: 243–250.
Simmons P, Beard B, Vehige J . Optimizing viscosity of ophthalmic solutions with the combination of two polymers. Presented at: 7th International Conference on the Tear Film & Ocular Surface: Basic Science and Clinical Relevance; September 18–21 2013 Taormina, Sicily, Italy.
Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL . Reliability and validity of the Ocular Surface Disease Index. Arch Ophthalmol 2000; 118: 615–621.
Methodologies to diagnose and monitor dry eye disease: report of the Diagnostic Methodology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf 2007; 5: 108–152.
Bron AJ, Evans VE, Smith JA . Grading of corneal and conjunctival staining in the context of other dry eye tests. Cornea 2003; 22: 640–650.
Chen JJ, Rao K, Pflugfelder SC . Corneal epithelial opacity in dysfunctional tear syndrome. Am J Ophthalmol 2009; 148: 376–382.
Le Henanff A, Giraudeau B, Baron G, Ravaud P . Quality of reporting of noninferiority and equivalence randomized trials. JAMA 2006; 295: 1147–1151.
Moore DB, Hammer JD, Akhtari R, Beck J, Sanders S, Kryscio RJ . Squeeze me if you can: variability in force requirements to extract a drop from common glaucoma bottles. J Glaucoma 2016; 25: 780–784.
Lee JH, Ahn HS, Kim EK, Kim TI . Efficacy of sodium hyaluronate and carboxymethylcellulose in treating mild to moderate dry eye disease. Cornea 2011; 30: 175–179.
Sanchez MA, Torralbo-Jimenez P, Giron N, de la Heras B, Herrero Vanrell R, Arriola-Villalobos P et al. Comparative analysis of carmellose 0.5% versus hyaluronate 0.15% in dry eye: a flow cytometric study. Cornea 2010; 29: 167–171.
Brignole F, Pisella PJ, Dupas B, Baeyens V, Baudouin C . Efficacy and safety of 0.18% sodium hyaluronate in patients with moderate dry eye syndrome and superficial keratitis. Graefes Arch Clin Exp Ophthalmol 2005; 243: 531–538.
Simmons PA, Liu H, Carlisle-Wilcox C, Vehige JG . Efficacy and safety of two new formulations of artificial tears in subjects with dry eye disease: a 3-month, multicenter, active-controlled, randomized trial. Clin Ophthalmol 2015; 9: 665–675.
Acknowledgements
The study was sponsored by Allergan plc (Dublin, Ireland). Writing and editorial assistance was provided to the authors by Michele Jacob, PhD, CMPP, of Evidence Scientific Solutions (Philadelphia, PA, USA), and funded by Allergan plc. All authors met the ICMJE authorship criteria. Neither honoraria nor payments were made for authorship.
Author Contributions
ML, FC and CB participated in the study design, data acquisition and interpretation, revised the manuscript critically for important intellectual content, and reviewed and approved the final version of the manuscript. AS and RL participated in the study design and data interpretation, revised the manuscript critically for important intellectual content, and reviewed and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
ML has served as a consultant for Allergan, Alcon, Bausch and Lomb, MSD, Santen/Novagali and Théa; AS and RL are employees of Allergan plc; CB has served as a consultant for Alcon, Allergan, Santen and Théa; and FC declares no conflict of interest. The funding body was involved in the design, data analysis and interpretation, revision of the manuscript for intellectual content, and decision to submit the manuscript for publication.
Additional information
This work was partially presented at the Congrès de la Société Française d’Ophtalmologie (SFO), May 7–10, 2016, Paris, France.
Supplementary Information accompanies this paper on Eye website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Labetoulle, M., Chiambaretta, F., Shirlaw, A. et al. Osmoprotectants, carboxymethylcellulose and hyaluronic acid multi-ingredient eye drop: a randomised controlled trial in moderate to severe dry eye. Eye 31, 1409–1416 (2017). https://doi.org/10.1038/eye.2017.73
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.73
This article is cited by
-
Allogeneic Serum Eye Drops: A Randomized Clinical Trial to Evaluate the Clinical Effectiveness of Two Drop Sizes
Ophthalmology and Therapy (2023)
-
A randomized multicenter clinical evaluation of sequential application of 0.3% and 0.15% hyaluronic acid for treatment of dry eye
Japanese Journal of Ophthalmology (2022)
-
A combination of CMC and α-MSH inhibited ROS activated NLRP3 inflammasome in hyperosmolarity stressed HCECs and scopolamine-induced dry eye rats
Scientific Reports (2021)
