
Abstract
Purpose
To describe the ocular and systemic phenotype in IQCB1-related disease.
Methods
Four cases (3 males, 1 female) with molecularly confirmed IQCB1-related disease underwent ophthalmological examination including best-corrected visual acuity (BCVA) measurement, fundus evaluation, electroretinography (ERG), and spectral-domain optical coherence tomography (SD-OCT). Systemic evaluation including abdominal ultrasound was performed in all cases.
Results
BCVA ranged from perception of light (Case-2; 1 year) to 20/125 (Case-1; 9 years). Fundus evaluation showed whitish or silvery reflex outside the vascular arcades in all cases; the reflex was circumferential, irregular and covered at-least 6 clock hours at younger ages (3 cases; 1–4 years). The reflex was less conspicuous with increasing age (Case-1 (9 years) and Case-4 (20 years)). The peripheral retinal SD-OCT scans showed evidence of extensive deposition at the level of retinal pigment epithelium with complete absence of overlying photoreceptor outer segments and myoid zone. The ERG was non-detectable in all cases. All cases harbored biallelic nonsense (p.R364*, p. R455*) or frameshifting (p.M370Yfs*49, p.C253Afs*9) mutations in IQCB1. Case-1 additionally had developmental delay, hemi-hyperplasia, toe syndactyly, and kidney cysts.
Conclusion
IQCB1-related syndromic or non-syndromic Leber congenital amaurosis (LCA) carries unique retinal characteristics which helps differentiate IQCB1-retinopathy from other genetic forms of LCA in childhood.
Similar content being viewed by others
Introduction
Primary cilia are non-motile sensory organelles that perform critical and diverse cell-specific functions in multiple mammalian cells during development and after birth.1, 2 In the retina, ciliary proteins modulate photoreceptor cilia length, and facilitate transport of membrane and soluble proteins along the cilium.2, 3 Mutations involving ciliary proteins commonly cause complex multi-system disorders broadly referred to as ciliopathies, a group of overlapping disorders that commonly manifest with retinal dystrophy, renal cysts, cardiac defects, digit anomalies, and cerebellar developmental defects.2 Senior-Loken syndrome (SLSN) is a rare, genetically heterogeneous, autosomal recessive ciliopathy that manifests with nephronophthisis (NPHP) and Leber Congenital Amaurosis (LCA).3, 4 Biallelic mutations in IQCB1 (NPHP5) cause SLSN and non-syndromic LCA.5, 6, 7
In this study, we report four cases harboring biallelic mutations in IQCB1 and describe the specific retinal phenotype best seen in childhood in this disorder; associated novel spectral-domain optical coherence tomography (SD-OCT) features are also documented.
Case reports
Case-1
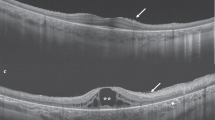
A male child of South-Asian descent was born (full-term) with right sided hemi-hypertrophy and bilateral toe syndactyly. His milestones were delayed, and the child developed nystagmus and symptoms of nyctalopia in infancy. At 4 years, best-corrected-distance-visual-acuity (BCVA) was 20/200 in either eye (Refraction: +5.25/+1.25 × 130° and +6.25 in the right and left eyes, respectively). Fundus examination showed a 270° ring of silvery white reflex in the periphery (Figure 1a, d, f, and h). Whitish dots were noted along the posterior edge of the reflex in some areas (Figure 1a, d, and f); lobular retinal pigment epithelial (RPE) changes were seen scattered anteriorly (Figure 1h). The SD-OCT showed relative preservation of the ellipsoid zone (EZ) in the central macula. The peripheral scans showed thick contiguous hyper-reflective deposition at the RPE corresponding to the areas of reflex; the photoreceptor outer structures [outer segments (OS), EZ and myoid zone] were absent in these regions, but the retina appeared thicker compared to adjacent regions (Figure 1b, c, e, and g). Full-field electroretinogram (ERG) showed non-detectable rod and cone responses. Ultrasound examination at 2 years of age showed bilateral renal parenchymal hyper-echogenicity. At 4 years, a cortical cyst was noted in the right kidney. The serum creatinine levels progressively worsened from 5 years of age; persistent hematuria was noted since 6 years of age. The child developed end-stage kidney disease that required renal transplant at 8 years.

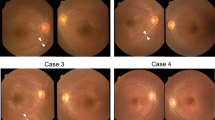
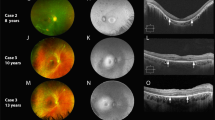
(Case-1): (a–h) Images obtained at initial presentation (4 years). All color fundus photos (a, d, f, and h) demonstrate peripheral whitish reflex/deposition. The 9 mm peripheral spectral-domain optical coherence tomography (SD-OCT) line scans (b, c, e, and g; Bioptigen) were obtained through regions of the whitish reflex and adjoining normal retina. All SD-OCT line scans demonstrate extensive hyper-reflective deposition at the level of retinal-pigment epithelium (RPE) corresponding to the whitish reflex or deposition. Note that the retina is thicker in regions of deposition despite the absence of photoreceptor outer-segments (OS) and myoid zone. The areas of normal color on fundus photography show absence of RPE deposition on SD-OCT; however, OS and myoid zone are absent even in these peripheral regions. (a) Right eye fundus photo showing mid-peripheral white reflex/deposits. The white lines correspond to the 9 mm Bioptigen spectral-domain optical coherence tomography (SD-OCT) line scans shown in (b) (top line) and (c) (bottom line), respectively. (d) Right eye superior-temporal fundus showing the mid-peripheral white reflex/deposits. The white line placed corresponds to the SD-OCT line scan shown in (e). (f) Left eye superior-temporal fundus photo showing the mid-peripheral white reflex/deposits; note that the pattern is symmetric between the two eyes. The white line shown in the figure corresponds to SD-OCT line scan shown in (g). (h) Left eye inferior-temporal fundus photo showing the mid-peripheral reflex/deposits along with lobular-pattern of RPE atrophy in the far periphery. (i–q) Images obtained at 9 years of age. (i) Right eye fundus photo showing mid-peripheral deep retinal hypo-pigmented regions reminiscent of early retinitis pigmentosa. The white line placed in the image correspond to 9 mm Cirrus SD-OCT line scan shown in (j). (j) SD-OCT image demonstrating the absence of the deposition earlier noted (b); there is also complete absence of OS and myoid zone. (k) Right eye fundus autofluorescence image showing peri-foveal ring of hyper-autofluorescence. There is an outer ring of hyper-autofluorescence in the temporal retina at the posterior edge of the atrophic changes. (l) A 9 mm SD-OCT line scan through the right macula demonstrating residual preservation of ellipsoid zone in the central macula. (m) Left eye fundus photograph showing deep retinal hypo-pigmented areas. The white lines are placed in the image corresponds to 9 mm SD-OCT images shown in (n) and (o). (n, o) SD-OCT image demonstrating the absence of OS and myoid zone, as well as the resolution of the RPE deposition previously noted (g). (p) Left eye fundus autofluorescence image showing peri-foveal ring of hyper-autofluorescence. The pattern is symmetric between the eyes. (q) SD-OCT line scan through the left macula demonstrating residual preservation of ellipsoid zone in the central macula.
At 9 years, the child developed photophobia; BCVA was 20/125 (Refraction: +2.00/+2.25 × 90° (either eye)). Fundus showed features of early retinitis pigmentosa (Figure 1i, m); the whitish reflex was scantily visible. The central sub-foveal thickness was 223 and 225 μm in the right and left eyes, respectively (Figure 1l, q); the peripheral scans failed to detect deposition from previously tested areas (Figure 1j, n, and o). Fundus auto-fluorescence (FAF) testing showed a peri-foveal ring of hyper-AF (Figure 1k, p). A homozygous stop mutation (c.1363C>T/p.R455*) was identified in IQCB1.7 Investigations for Beckwith Wiedemann related hemi-hypertrophy syndromes (methylation abnormalities of 11p15) were negative.
Case-2
A 1-year-old female child of Chinese descent presented with nystagmus, oculo-digital reflex and history of staring towards bright light. Visual acuity was perception of light (Refraction: +5.00 (either eye)). Retinal examination showed a peripheral band (300°) of whitish deep retinal reflex (Figure 2a, d, f, and h). The SD-OCT showed residual preservation of EZ in the central macula (Figure 2c). The peripheral scans showed thick, contiguous deposition at the RPE corresponding to the whitish reflex, and photoreceptor OS and myoid zone were absent (Figure 2b, e, g, and i). The ERG was non-detectable to all tested stimuli. Detailed renal function tests and kidney ultrasound were normal. A homozygous null mutation (c.1060C>T/p.R364*) was identified in IQCB1.8, 9

(a–i) Case-2. The color fundus photos (a, d, f, and h) demonstrate mid-peripheral whitish reflex. The 9 mm peripheral spectral-domain optical coherence tomography (SD-OCT) line scans (b, e, g, and i; Bioptigen) were obtained through regions of the whitish reflex and adjoining normal retina. All peripheral SD-OCT line scans (b, e, g, and i) demonstrate extensive contiguous hyper-reflective deposition at the level of retinal-pigment epithelium (RPE) corresponding to the deposition. The areas of normal color on fundus photography show absence of RPE deposition on SD-OCT. Note that there is absence of photoreceptor outer-segments (OS) and myoid zone in the peripheral retina. The retina appears thicker in the regions of the deposition in some scans (i). (a) Right eye superior-temporal quadrant fundus photo showing mid-peripheral white reflex. The white line corresponds to SD-OCT line scans shown in (b). (c) SD-OCT line scan through the right macula showing residual preservation of ellipsoid zone in the central macula. (d) Right eye temporal fundus photograph showing the reflex. The white line placed corresponds to SD-OCT scan shown in (e). (f) Left eye fundus photo showing the mid-peripheral white reflex. The white line placed corresponds to SD-OCT scan shown in (g). (h) Left eye fundus photo showing the mid-peripheral white reflex. The white line placed corresponds to SD-OCT scan shown in (i). (j–q) Case-3. The color fundus photos (j, l, n, and p) demonstrate mid-peripheral greyish-white reflex. The 9 mm peripheral SD-OCT line scans (m and q; Bioptigen) were obtained through regions of the grayish-white reflex; both scans (m, q) demonstrate extensive contiguous hyper-reflective deposition at the level of RPE corresponding to the reflex. Note that there is absence of photoreceptor OS and myoid zone in the peripheral retina. (j) Right eye temporal fundus photo showing the mid-peripheral grayish-white reflex. The white line placed corresponds to SD-OCT scan shown in (k). (k) SD-OCT line scan through the right macula showing some residual preservation of OS and ellipsoid zone in the central macula. At the temporal end, hyper-reflective deposition at the level of RPE corresponding to the grayish-whitish reflex in the fundus is noted. Outside the central macula, there is absence of OS and myoid zone. (l) Right eye superior-temporal fundus photo showing the grayish-white reflex. The white line placed corresponds to SD-OCT scan shown in (m). (n) Left eye temporal fundus photo showing the grayish-white reflex. (o) SD-OCT line scan through the left macula showing some residual preservation of OS in the central macula. (p) Left eye superior-temporal fundus photo showing the grayish-white reflex. The white line placed corresponds to SD-OCT scan shown in (q). (r–w) Case 4. (r) Right eye fundus photograph showing disc drusen and para-foveal yellowish reflex; the mid-peripheral retina shows multiple deep retinal hypopigmented areas as seen in early retinitis pigmentosa. (s) SD-OCT line scan through the right macula; there is complete disruption of the OS, ellipsoid zone and myoid zone in the macula. There is some evidence of localized hyper-reflective RPE deposition. (t, u) SD-OCT line scans through regions marked in the fundus photo (v). Note the complete absence of OS and myoid zone throughout the scans. Also, there is absence of RPE deposition. (v) Left eye temporal fundus photo showing silvery reflex along the supero-temporal arcade with some lobular architecture to it. The white lines placed represent (t) (top line) and (u) (bottom line) respectively. (w) Left eye fundus autofluorescence showing foveolar hyper-autofluorescence; surrounding this there are alternate rings of hypo- and hyper-autofluorescence.
Case-3
A 1-year-old male child of Lebanese descent presented with history of nystagmus, oculo-digital reflex and staring at bright light. Visual acuity was perception of light (Refraction: +10.00; both eyes). Retinal evaluation showed greyish-white deep retinal reflex in the mid-periphery involving superior and temporal quadrants (Figure 2j, l, n, and p). The SD-OCT showed residual preservation of EZ in the central macula (Figure 2k, o). The peripheral scans showed thick, contiguous deposition at the RPE; photoreceptor OS and myoid zone were absent in these regions (Figure 2m, q). The ERG was non-detectable to all stimuli. Renal function tests and ultrasound were normal. A novel homozygous frameshifting insertion (c.1107dupT/p.M370Yfs*49) was identified in IQCB1.
Case-4
A 20-year-old male of Filipino descent presented with history of infantile onset nystagmus, photophobia and poor vision. The patient had nyctalopia since childhood. BCVA was 20/500 in either eye. Retinal examination showed a para-foveal annular yellowish reflex (2r); mid-peripheral retina showed patchy retinal silvery reflex with patchy lobular architecture (2v). The macular OCT showed disruption of EZ and OS in the sub-foveal region (2s). Some macular line scans showed localized evidence of RPE deposition (Figure 2s); peripheral scans showed absence of photoreceptor OS and myoid zone, and there was no evidence of RPE deposition (Figure 2t, u). FAF showed foveal hyper-autofluorescence surrounded by a ring of hypo-autofluorescence (Figure 2w). The ERG was non-detectable to all stimuli. Renal function tests and abdominal ultrasound were normal. Compound heterozygous mutations (c.1060C>T/p.R364*; c.757delT/p.C253Afs*9) were identified in IQCB1; the frame shifting variant is novel.
Discussion
This report highlights the fundus and peripheral SD-OCT features in IQCB1-related retinopathy. The whitish/silvery deep retinal reflex was observed in all cases; however, it was more conspicuous in the peripheral retina of young affected individuals. A previously described lobular aspect to the retinal changes was observed in patches in two of our cases (Case-1 at 4 years; Case-4 at 20 years).6 The peripheral OCT findings are novel and suggest the origins of the fundus reflex to be consequent upon the extensive deposition at the level of the RPE. There is absence of photoreceptor OS and myoid zone overlying the deposition, and in the peripheral retina. The central macula showed preservation of EZ in younger affected individuals as reported previously.10, 11
Mice knock out models of Nphp5−/− demonstrated absence of development of rod OS and axoneme with ensuing nuclear degeneration.12 A spontaneously occurring dog model of Nphp5 showed 90% of cones to have abnormal OS development, but there was some preservation of OS in the cone-rich visual streak.10 Hence, the widespread absence of OS in the peripheral retina along with residual preservation of foveal cones in humans shows some similarity to both mice and dog models, respectively.10, 12 The peripheral retinal deposition may represent abnormally developed photoreceptor OS; however, no deposition was observed in animal models.10, 12 Secondary changes likely explain the paucity of retinal deposition in older patients (Case-1 at 9 years and Case-4 at 20 years).
Hyperopic refractive error and the classical triad of LCA (nystagmus, poor vision and non-detectable ERG) seen in this series were common amongst previously reported IQCB1 cases.6, 7 The p.R455* variant manifesting as SLSN in the present study, has previously been associated with non-syndromic LCA (6 years).7 The p.R364* variant manifesting as non-syndromic LCA in this study (1 year and 20 years, respectively), has been previously associated with LCA (28 years) and SLSN (16 years and 12 years).8, 9 Hence, there are likely genetic or environmental modifiers that influence IQCB1-phenotype. Syndactyly and developmental delay commonly seen in Bardet–Biedl syndrome was observed in one of our cases, and is consistent with the phenotypic overlap described in ciliopathies.13, 14 Hemi-hypertrophy seen in one case in the present study has not been associated with ciliopathy; whether this is specific to IQCB1 needs further evidence.
Multimodal phenotyping of retinal dystrophies, even at a young age, guide genetic diagnosis and shed light on underlying pathology. This report identifies a whitish peripheral retinal reflex due to deep retinal deposition as a phenotypic marker for IQCB1-retinopathy.

References
Satir P, Pedersen LB, Christensen ST . The primary cilium at a glance. J Cell Sci 2010; 123 (4): 499–503.
Rachel RA, Li T, Swaroop A . Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia 2012; 1 (1): 22.
Loken AC, Hanssen O, Halvorsen S, Jolster NJ . Hereditary renal dysplasia and blindness. Acta Paediatr 1961; 50: 177–184.
Senior B, Friedmann AI, Braudo JL . Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol 1961; 52: 625–633.
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 2005; 37 (3): 282–288.
Estrada-Cuzcano A, Koenekoop RK, Coppieters F, Kohl S, Lopez I, Collin RW et al. IQCB1 mutations in patients with leber congenital amaurosis. Investig Ophthalmol Vis Sci 2011; 52 (2): 834–839.
Stone EM, Cideciyan AV, Aleman TS, Scheetz TE, Sumaroka A, Ehlinger MA et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol 2011; 129 (1): 81–87.
Chen Y, Zhang Q, Shen T, Xiao X, Li S, Guan L et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Investig Ophthalmol Vis Sci 2013; 54 (6): 4351–4357.
Tong H, Yue Z, Sun L, Chen H, Wang W, Wang H . Clinical features and mutation of NPHP5 in two Chinese siblings with Senior-Loken syndrome. Nephrology (Carlton) 2013; 18 (12): 838–842.
Downs LM, Scott EM, Cideciyan AV, Iwabe S, Dufour V, Gardiner KL et al. Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum Mol Genet 2016; 25 (19): 4211–4226.
Cideciyan AV, Rachel RA, Aleman TS, Swider M, Schwartz SB, Sumaroka A et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet 2011; 20 (7): 1411–1423.
Ronquillo CC, Hanke-Gogokhia C, Revelo MP, Frederick JM, Jiang L, Baehr W . Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation. FASEB J 2016; 30 (10): 3400–3412.
Deveault C, Billingsley G, Duncan JL, Bin J, Theal R, Vincent A et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat 2011; 32 (6): 610–619.
Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, Ibrahim Khalil M et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet 2014; 94 (1): 73–79.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Vincent, A., AlAli, A., MacDonald, H. et al. Specific retinal phenotype in early IQCB1-related disease. Eye 32, 646–651 (2018). https://doi.org/10.1038/eye.2017.283
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.283
