
Abstract
The past two decades has been an amazing time in the advancement of cancer treatment. Molecularly targeted therapy is a concept in which specific cellular molecules (overexpressed, mutationally activated, or selectively expressed proteins) are manipulated in an advantageous manner to decrease the transformation, proliferation, and/or survival of cancer cells. In addition, increased knowledge of the role of the immune system in carcinogenesis has led to the development of immune checkpoint inhibitors to restore and enhance cellular-mediated antitumor immunity. The United States Food and Drug Administration approval of the chimeric monoclonal antibody (mAb) rituximab in 1997 for the treatment of B cell non-Hodgkin lymphoma ushered in a new era of targeted therapy for cancer. A year later, trastuzumab, a humanized mAb, was approved for patients with breast cancer. In 2001, imatinib was the first small-molecule kinase inhibitor approved. The approval of ipilimumab—the first in class immune checkpoint inhibitor—in 2011 serves as a landmark period of time in the resurgence of immunotherapy for cancer. Despite the notion that increased tumor specificity results in decreased complications, toxicity remains a major hurdle in the development and implementation of many of the targeted anticancer drugs. This article will provide an overview of the current cellular and immunological understanding of cancer pathogenesis—the foundation upon which molecularly targeted therapies were developed—and a description of the ocular and neuro-ophthalmic toxicity profile of mAbs, immune checkpoint inhibitors, and small-molecule kinase inhibitors.
Similar content being viewed by others

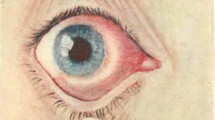
Introduction
War is a recurrent and unfortunate record in the history of human civilization that has culminated in indescribable violence and unspeakable death. However, amazingly within the confines of war have risen some of the greatest advancements in medicine. It is within this setting—in particular World War II with the study of mustard gas—that the annals of cancer chemotherapy began touching the lives of millions of people. It is estimated that in 2016, over 1.6 million people in the United States will be diagnosed with cancer and over a half a million will die.1 The amount of money being spent on research and development of new cancer therapies is staggering with a record $43 billion dollars spent in 2014. Nearly 30% of all registered clinical trials on the clinicaltrials.gov website pertain to cancer drugs. Such large numbers emphasize the urgency of finding a cure for cancer.
In the context of co-morbid systemic diseases and patient expectations, the oncologist has a wide variety of treatment options to choose from based on the histological type, molecular marker, and clinical stage of cancer (Table 1). Since its first clinical application in the early 1940s, cytotoxic chemotherapy has been the mainstay of medical treatment for cancer. However, in the past two decades treatment options have expanded dramatically for many cancers, allowing oncologists to provide an increasingly personalized approach.2 Much has been learned about normal cell development, differentiation, survival, proliferation, and ultimate death; which has in turn increased our knowledge and understanding of carcinogenesis. However, there is still much that is not understood about the epigenetic mechanisms in cellular transformation to immortality and the complicated interplay between the immune system and cellular regulation. It should also be kept in mind that the financial impact of targeted cancer therapies has been enormous both in terms of sales (profit) and health care cost.3
Efficacy is a major goal in cancer drug development. However, safety and toxicity often lead to either limited use of a drug or prevent the utilization of a drug in clinical practice. Despite a very arduous, comprehensive and costly process of drug research and development culminating in approval by the United States Food and Drug Administration (FDA),4 the reporting of adverse events (AEs) from clinical trials and other databases remains inconsistent and often difficult to interpret.5, 6 In a study that reviewed the results of randomized control trials (RCTs) and updated package inserts (drug label) of targeted cancer drugs, nearly 40% of serious AEs were not published in the initial RCT paper and ~50% of the serious AEs were not included in the initial package insert.7 The financial and medical burden of cancer drug toxicity and AEs is enormous.8 In many cases it can be difficult to ascertain the true cause and effect of an AE because of its rarity and the fact that there may be confounding factors—such as the combination of medical or radiation therapy—in the treatment regimen of patients.
This review article aims to provide an overview of the current cellular and immunological understanding of carcinogenesis— the foundation upon which molecularly targeted therapies were developed—within the framework of discussing the ocular and neuro-ophthalmic toxicity profile of selected monoclonal antibodies (mAbs), immune checkpoint inhibitors, and small-molecule kinase inhibitors.
Molecularly targeted therapy
The approval of the first targeted mAb—rituxamb— in 1997 was the clinical starting point for a new generation of molecularly targeted therapy in cancer.9 Aside from surface receptors, other targets for this novel approach include intracellular signal proteins and metabolic molecules. Targeted therapy not only affects tumor cells but also immune cells (ie, T and B cells) and vascular-stromal cells.2, 10
Monoclonal antibodies
The clinical development of mAbs began in earnest in 1972 when Kohler and Meilstein published their ground breaking technique of producing murine mAbs.11 This was followed in 1987 with the generation of a chimeric mAb.12 With the use of XenoMouse technology, panitumumab was the first human mAb approved for use in 2006.13 A variety of mAbs exist including uncongugated (naked), conjugated (attached to effector molecules such as cytotoxic drug, bacterial or plant toxin, or radiopharmaceutical agents),14 and bispecific.15 Other antibody products include antibody-ligand fusion proteins16 and immunoliposomes.14 The nomenclature of mAbs follows a very specific characterization scheme.17 The elements that make up an antibody name are prefix+target/disease class infix+source infix+stem. Some commonly used target/disease class infix’s include -tu/-t for tumors, -li/-l for immunomodulatory, and -ba/-b for bacterial. The source infix can be –zu for humanized, -o for mouse, -u for fully human, or -xi for chimeric. The stem is either –mab for monoclonal or –pab for polyclonal.
The use of mAbs has become one of the cornerstone treatments in the fight against cancer. There are currently14 FDA-approved mAbs (not including the immune checkpoint inhibitors) on the market (Table 2).18 It has been estimated that by the year 2020 the world-wide sales of mAbs will be approximately $125 billion.19
The mechanism of action (MOA) by which mAbs exert their anticancer effects is varied and includes apoptosis, activation or inhibition of a surface cell receptor, antibody-dependant cellular cytotoxicity (ADCC), and complement-dependant cytotoxicity (CDC). In addition mAbs can target tumor cells, immune cells (ie, T-cell or B cell) or vascular/stromal cells.16, 20, 21
The toxicity or AEs of a particular mAb is primarily related to its MOA and/or the unintended targeting of a cell or organ system.22 Toxicity can be limited if the mAb is directed at a specific target on the neoplastic cell without affecting normal or healthy cells. The four general categories of mAb associated AEs are immune reaction (infusion reaction), excessive cytokine release or storm, immunosuppression, and autoimmunity.22, 23
In general terms, the ocular toxicity profile of mAbs is good. Several review papers have summarized the ocular AEs reported with mAbs.24, 25, 26, 27, 28 Neurological side effects due to demyelination from mAbs have been associated with antitumor necrosis factor agents, none of which are approved for cancer therapy.29 The following is a focused discussion of the ocular and neuro-ophthalmic complications associated with several selected mAbs.
Alemtuzumab
Alemtuzumab is a humanized IgG1 mAb that targets a variety of immune cells including B cells, T cells, and macrophages that express the cluster of differentiation (CD) 52 antigen. No specific ocular toxicities have been associated with alemtuzumab.
A unique AE associated with alemtuzumab is the increase in autoimmunity. The black box of the package insert indicates the fatal risks of pancytopenia/marrow hypoplasia, autoimmune idiopathic thrombocytopenia, and autoimmune hemolytic anemia.30 Post-marketing experience has identified several autoimmune disorders including Goodpasture syndrome, Graves disease, aplastic anemia, Guillain-Barré syndrome, and chronic inflammatory demyelinating polyradiculoneuropathy. The risk of Graves disease in cancer patients receiving alemtuzumab is not known but in multiple sclerosis (MS) patients it has been estimated to be ~20%.31 In the phase 2 clinical trial for MS, of the 39 patients that developed Graves disease, 4 patients developed thyroid eye disease.32 Progressive multifocal leukoencephalopathy (PML) has been reported to occur in the setting of alemtuzumab therapy (see section on ritixumab below).33, 34
Bevacizumab
Bevacizumab, a humanized IgG1 mAb, targets vascular endothelial growth factor (VEGF) thereby preventing binding to the VEGF receptor (VEGFR), resulting in the inhibition and regression of the tumor vasculature.35 Sherman et al, reported 6 patients who developed optic neuropathy while taking bevacizumab in addition to fractionated radiation therapy and temozolomide for glioblastoma.36 In a meta-analysis, bevacizumab was found to be associated with a 3 fold higher risk of cerebral stroke and hemorrhage.37 Posterior reversible encephalopathy syndrome (PRES), a disorder of cerebral vascular autoregulation resulting in vasogenic edema, has been reported with bevacizumab.38
Brentuximab vedotin
Brentuximab vedotin is a conjugated mAb against the cell surface protein CD30, used in Hodgkin lymphoma and anaplastic lymphoma, that has been associated with peripheral sensory neuropathy and PML.39 There are no known ocular toxicities.
Cetuximab
Cetuximab is a chimeric IgG1 mAb that is an epidermal growth factor receptor (EGFR) inhibitor. EGFR is found in corneal epithelial cells and hair follicles, therefore the ocular side effects of cetuximab are related to the function of the inhibition of the receptor thereby causing keratitis, conjunctivitis, blepharitis and eyelash trichomegaly.25, 26, 28
Rituximab
Rituximab is a chimeric IgG1 mAb directed against CD 20, which is predominately found on B cells. Its MOA is not entirely known but is thought to work by ADC, ADCC and apoptosis. The package insert carries a black box warning regarding fatal infusion reactions, hepatitis B virus reactivation, severe mucocutaneous reactions and PML. PML is an infectious demyelinating disease of the central nervous system caused by the polyomavirus John Cunningham (JC) virus associated with significant morbidity and mortality.40 From an ophthalmic perspective patients who develop PML develop visual complaints and visual field defects often in a homonymous heminaopsia pattern due to involvement of the posterior visual pathways within the parietal–occipital lobes. The exact pathophysiology is not known but it is believed to be due to the re-population of immature B cells that contain the JC virus.41 Nearly a decade after receiving approval, in 2006 the FDA disseminated an alert to physicians regarding 2 patients with systemic lupus erythematosus (SLE) who developed PML following rituximab treatment.42 The manufacturer of rituximab sent out 2 letters to physicians in 2008 describing the association of rituximab with PML.41 Subsequently the package insert was modified with the addition of the risk of PML and the institution of a risk evaluation and mitigation strategy (REMS) plan.43 The package insert indicates that the majority of patients developed PML within 12 months from the last infusion and that many of the patients were concomitantly treated with chemotherapy or a hematopoietic stem cell transplant.44 The exact risk of developing PML in the setting of cancer treatment is not known, but the overall frequency has been estimated to be 1 : 30 000.34
Aside from rituximab, alemtuzumab and brentuximab vedotin the other mAbs associated with PML include bevacizumab, cetuximab, and ibritumomab tiuxetan.45, 46
Trastuzumab
Trastuzumab is a humanized IgG1 mAb that targets the epidermal growth factor receptor 2 (Her2).47 Dry eye, tearing, conjunctivitis and blurred vision have been reported with trastuzumab.25 Saleh et al described a unique case of bilateral macular ischemia and edema in the setting of trastuzumab. However, the patient also received radiation and docetaxel therapy.48 In the review by Huillard et al,26 papilledema, retinal hemorrhage, retinal artery occlusion, and retinal vein occlusion have been reported with trastuzumab.
Treatment
The decision to discontinue or continue the offending mAb needs to be discussed with the prescribing oncologist and depends on the severity of the AE. In some instances it may be possible to treat the symptoms either with supportive therapy or more directed therapy without discontinuing the mAB.
PML can result in severe neurological deficits and is associated with a high mortality rate. Re-institution of normal immune system function is critical to suppress the infection. Plasma exchange has been advocated but the efficacy remains debated.49 Other treatments implemented for PML include mirtazapine, mefloquin, cytabrine, and cidofir but none of them at this time have been proven to be effective.50
The mainstay of treatment for PRES is supportive and directed at symptom management (ie, anti-seizure medication, anti-hypertensive medication and correction of electrolyte abnormalities). In some cases re-challenging with the offending cytotoxic agent has not resulted in a recurrence of PRES, but such a re-challenge with mAbs has not been tried.51
Immune checkpoint inhibitors
Cancer immunotherapy relies on the strategy of actively manipulating one of the three basic steps in the generation and regulation of antitumor immunity (Figure 1); and historically had been limited by a lack of understanding of immunoregulatory mechanisms.52 Immune checkpoint inhibitors are fundamentally different from all the other targeted therapies because of their unique MOA. Instead of a passive or an immunomodulatory role, immune checkpoint inhibitors activate the immune system by blocking the immune inhibitory pathways activated by cancer cells (Figure 2).52

Generation and regulation of antitumor immunity. Understanding the events in generating and regulating antitumor immunity suggests at least three sites for therapeutic intervention: promoting the antigen presentation functions of dendritic cells, promoting the production of protective T-cell responses and overcoming immunosuppression in the tumor bed. Antitumor immune responses must begin with the capture of tumor associated antigens by dendritic cells, either delivered exogenously or captured from dead or dying tumor cells. The dendritic cells process the captured antigen for presentation or cross-presentation on MHC class II and class I molecules, respectively, and migrate to draining lymph nodes. If capture and presentation occurred in the presence of an immunogenic maturation stimulus, dendritic cells will elicit anticancer effector T-cell responses in the lymph node; if no such stimulus was received, dendritic cells will instead induce tolerance leading to T-cell deletion, anergy or the production of Treg cells. In the lymph node, antigen presentation to T cells will elicit a response depending on the type of dendritic cell maturation stimulus received and on the interaction of T-cell co-stimulatory molecules with their surface receptors on dendritic cells. Thus, interaction of CD28 orOX40 with CD80/86 orOX40L will promote potentially protective T-cell responses, while interaction of CTLA4 with CD80/86 or PD-1 with PD-L1/PD-L2 will suppress T-cell responses, and possibly promote Treg formation. Antigen-educated T cells (along with B cells and NK cells) will exit the lymph node and enter the tumor bed, where a host of immunosuppressive defense mechanisms can be produced by tumors (or infiltrating myeloid cells) that oppose effector T-cell function. These include the upregulation of PD-L1/L2 on the cancer cell surface, release of PGE2, arginase and IDO (all T-cell suppressors), and the release of VEGF (triggered in part by intratumoral hypoxia), which inhibits T-cell diapedesis from the vasculature, and thus infiltration into the tumor bed. (Reprinted with permission: Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011 Dec 21; 480(7378): 480–489). A full color version of this figure is available at Eye online.

Site of immune checkpoint inhibitor action. Anti-CTLA4 prevents binding of CTLA4 to CD80 and CD86 ligands expressed on the surface of dendritic cells. The binding of CD28 to CD80 and CD86 ligands on the APC is a second co-stimulatory signal. CTLA-4 competes with CD28 in binding for CD80 and CD86 ligands. PD-L1 binds to PD-1 thereby de-activating T cells. Blocking either PD-L1 or PD-1 on cancer cells results in the activation of T cells. Anti-CTLA 4 action occurs in the lymph nodes therefore earlier on in the immune response, as compared to anti-PD-1, which is critical in the tumor microenvironment. APC, antigen presenting cell; CD, cluster of differentiation; MHC, major histocompatibility complex; PD, programmed death; PD-L, programmed death ligand; TCR, T-cell receptor. (Illustration by Rob Flewell, CMI).
There are currently four FDA-approved immune checkpoint inhibitors that activate the immune response through distinct mechanisms (Table 2). Ipilimumab is a human mAb against the cytotoxic T lymphocyte antigen 4 (CTLA4), which normally serves to modulate T-cell activity in the lymph node in response to T-cell activation, competitively binding CD 80 and CD 86 ligands on dendritic cells thereby limiting persistent activation. By blocking this interaction, ipilimumab allows for continued T-cell proliferation thus inhibiting an immune ‘checkpoint’. In comparison, nivolumab and pembrolizumab target programmed death-1 (PD-1) in the tumor microenvironment. The interaction with PD-1 and the programmed death-ligand 1 (PD-L1) functions to limit T-cell expansion and prevent tissue damage from inflammation. Certain cancers have co-opted this mechanism by expressing high levels of PD-L1, effectively evading immune detection. The anti-PD-1 antibodies release this inhibitory blockade, allowing for continued T-cell activation, with the potential for remarkable clinical responses across a wide variety of tumor types. Atezolizumab targets PD-L1 resulting in a similar immune activation (removal of inhibition) effect as nivolumab and pembrolizumab.
Immune checkpoint inhibitors have a unique safety profile because the immune system is activated, with subsequent autoimmune toxicity termed immune-related adverse events (IRAEs).53 IRAEs are common, occurring in 70–90% of patients and affect multiple organ systems including the skin, gastrointestinal tract, lung, kidney, blood, adrenal gland, and thyroid.53, 54 In addition, immune checkpoint inhibitors have been associated with autoimmune diseases such as SLE, rheumatoid arthritis, Graves disease, Vogt–Koyanagi–Harada (VKH) syndrome, myasthenia gravis, MS and giant cell arteritis.27, 53, 55, 56, 57
Ipilimumab
As the first approved immune checkpoint inhibitor, with the largest amount of published data, the neuro-ophthalmic side effects of ipilimumab will be emphasized in this section.
The incidence of ocular IRAEs is <1%.58 Several ophthalmic toxicities have been associated with the use of ipilimumab including blepharitis, conjunctivitis, keratitis, episcleritis, scleritis, uveitis, serous retinal detachment, and choroidal neovascularization.24, 26, 28, 58, 59
Yeh and Francis reported bilateral optic nerve edema subretinal fluid and anterior uveitis in a 67-year-old man treated with ipilimumab for metastatic melanoma. Cranial magnetic resonance imaging (MRI) revealed small vessel ischemic changes and a lumbar puncture documented a slightly elevated intracranial pressure (23.5 cmH2O). Only topical steroids were given with continuation of the ipilimumab and by four months the optic nerve edema resolved but was replaced with pallor associated with persistent bilateral visual field defects.60 Papillitis in the setting of VKH and thyroid eye disease due to Graves disease have been described with ipilimumab.27, 28, 58 Hahn and Pepple reported a patient with bilateral iritis and bilateral optic nerve edema associated with macular edema, which they termed neuroretinitis.61 Several case reports and case series have documented the occurrence of orbital inflammation.28, 62, 63 Johnson et al56 described a 69-year-old woman with ptosis and external ophthalmoparesis due to myasthenia gravis after receiving ipilimumab infusions.
Neurological IRAEs associated with ipilimumab are diverse including both the central and peripheral nervous system. Hypophysitis, PRES, peripheral neuritis (Guillain-Barré syndrome), meningitis, encephalitis, myelitis and facial neuritis have all been reported with ipilimumab treatment.28, 53, 54, 64 Most recently Gerdes et al described a case of a 29-year-old man treated with ipilimumab for metastatic melanoma who prior to treatment had a cerebral MRI that showed white matter T2 hyperintensities consistent with a radiologically isolated syndrome. During treatment the patient developed hypophysitis. Four months after the first infusion the patient developed a clinical attack of demyelination (thermhypesthesia of both feet). After the second infusion, a repeat MRI of the brain showed increased lesions resulting in a brain biopsy consistent with active MS; and then 3 months after the last infusion the patient developed optic neuritis of the left eye.57
Pembrolizumab
Roberts et al recently described a patient who was treated with pembrolizumab for cutaneous metatstatic melanoma and developed chorioretinal scars and pigment clumping in the peripheral retina of both eyes. The serum was positive for multiple autoretinal antibodies. The authors postulated that pembrolizumab may have induced (in their words ‘unleased’) an autoimmunity state in the patient leading to the fundus findings in the presence of melanoma-associated retinopathy.65
Treatment
Established treatment guidelines have been published for the more common IRAEs involving the skin, gastrointestinal tract, endocrine system and lung but not for the eye or nervous system.66 In general, for mild IRAEs, continuation of the medication with supportive therapy can be an effective strategy.54, 67 However, for more severe cases corticosteroids (either oral or intravenous) are the primary treatment modality. In those cases that are, either recalcitrant to corticosteroids or recurrence of the IRAE occurs during the tapering of the corticosteroids, infliximab, intravenous immunoglobulin (IVIG) and plasma exchange is recommended.54, 66 Wilson et al described a patient on ipilimumab who despite systemic corticosteroids developed recurrent visual loss due to optic neuritis requiring plasma exchange and mycophenolate mofetil.68 In a case report of autoimmune encephalitis, the patient received rituximab in addition to corticosteroids and IVIg.69 Occasionally, immune checkpoint inhibitor induced myasthenia gravis may spontaneously resolve but in most cases require an anti-acetylcholinesterase and/or corticosteroids and in severe cases IVIg or plasma exchange.70
Small-molecule kinase inhibitors
It is beyond the scope of this review article to discuss the intricacies and complexities of the various intracellular signal molecules and their relationship to one another in maintaining normal cell development, differentiation, survival and proliferation. Suffice it to say that dysfunction of these signal pathways results in an abnormal cell cycle and development of neoplasia. A basic knowledge of signal transduction dysregulation of cancer cells is important in the context of understanding the efficacy and toxicity of small-molecule kinase inhibitors.71 Although incompletely understood, it is apparent that there are a number of complex, independent, parallel and interconnected signal transduction pathways involving the extracellular, cell surface and intracellular compartments (Figure 3).

Rationale for targeting both the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways for suppressing cancer growth. (a) The Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways are both activated by upstream receptor ligation and frequently co-regulate many downstream targets in parallel. Thus for effective elimination of many cancers or prevention of aging, it may be necessary to target both signaling pathways. Activation of these pathways could also result in increased transcription of many genes that promote cellular growth and malignant transformation. (b) Inhibition of mTOR can result in the induction of autophagy, which is a very important mechanism of cell death, especially in solid tumors. (c) As described previously, both the Ras/Raf/MEK/ERK and Ras/PI3K/ PTEN/Akt/mTOR pathways regulate the activity of apoptotic proteins by post-translational mechanisms. Targeting this pathway may also contribute to the induction of apoptosis. Signaling molecules promoting phosphorylation events are indicated in green. Stimulatory signaling events are indicted in green lines with a green arrow before the target of the phophorylation. Small-molecule inhibitors are indicated in red. Inhibitory phosphorylation events are indicated in red lines with a block on the end before the target of the inhibition. Inhibitory signaling or proapoptotic molecules or inactivated molecules are indicated in yellow. A growth factor and a growth factor receptor are indicated in purple. Active transcription factors are indicated in purple diamonds. Inactivated transcription factors are indicated in yellow diamonds. (Reproduced with permission: Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Laidler P, Milella M, Tafuri A, Bonati A, Evangelisti C, Cocco L, Martelli AM, McCubrey JA. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011 Mar; 2(3): 135–164). A full color version of this figure is available at Eye online.
Small-molecule kinase inhibitors are a group of anticancer drugs that affect the intracellular signal pathways that are dysfunctional in cancer cells.72, 73 Small-molecule kinase inhibitors are adenosine triphosphate (ATP) mimetics or analogues that can target the extracellular or intracellular components of a cell surface receptor as well as intracellular protein kinases.74 As a result of this inhibition of ATP transfer there is no post-translational phosphorylation, causing inactivity of the receptor or molecule leading to inactivity or paradoxical hyperactivity of downstream signal pathways.75
Small-molecule kinase inhibitors are distinct from mAbs because they are smaller in size, shorter in half-life, administered orally, metabolized by the cytochrome p450 enzymes and have a different MOA (Figure 4).2 As is the case for mAbs, the nomenclature for identifying a small-molecule kinase inhibitor follows a specific scheme. All small-molecule kinase inhibitors end with the suffix –ib, and the stem -tin indicates a tyrosine kinase inhibitor.76 FDA approval of the first small-molecule kinase inhibitor—imatinib—occurred 15 years ago.77 Since then there has been a steady increase in the number of drugs that have reached the market, totaling 30 as of 2016 (Table 2).78 Some of the kinase inhibitors target a single protein while others target multiple proteins.74 The following section will be discuss the small-molecule kinase inhibitors as a group based on their target molecule with special emphasis on a particular drug if it has been associated with a unique or noteworthy toxicity.

Distinct mechanisms of small-molecule inhibitors and monoclonal antibodies for targeting receptor tyrosine kinases in cancer cells. (a). Epidermal growth factor receptor (EGFR) and receptor tyrosine kinase (RTK)-dependent growth signaling in cancer cells. The extracellular region of EGFR consists of four domains, the ligand-binding domains (L1 and L2) and the cysteine-rich domains (CR1 and CR2), and the C-terminal domain of EGFR contains six tyrosine residues (Y; only two are depicted here for simplicity). Following the activation of EGFR by ligand binding or ligand-independent dimerization, the Ras–Raf–MEK–MAPK pathway is activated through the growth factor receptor bound protein 2 (GRB2)–SOS complex. EGFR-mediated signaling also activates the phosphatidylinositol 3-kinase (PI3K)– AKT pathway, which contributes to anti-apoptotic effects of EGFR activation. In addition, signal transducer and activator of transcription (Stat) proteins (STAT1, STAT3, and STAT5) are also activated. The coordinated effects of these EGFR downstream signaling pathways lead to the induction of cellular responses including proliferation, differentiation, cell motility, adhesion, and angiogenesis. The deregulation of EGFR-mediated signaling in some cancer cells leads to aberrant proliferation, invasion, metastasis, and neovascularization. (b) Small-molecule tyrosine kinase inhibitors (TKIs) such as gefitinib function as ATP analogues and inhibit EGFR signaling by competing with ATP binding within the catalytic kinase domain of RTKs. As a result, the activation of various downstream signaling pathways is blocked. Each TKI has a different selectivity for RTKs, and some are dual- or multi-selective, which might provide a therapeutic advantage. (c) By contrast, therapeutic monoclonal antibodies (mAbs) bind to the ectodomain of the RTK with high specificity (for example, cetuximab binds to the L2 domain of EGFR, and thereby inhibits its downstream signaling by triggering receptor internalization and hindering ligand–receptor interaction. Unlike small-molecule inhibitors, mAbs also activate Fcγ- receptor-dependent phagocytosis or cytolysis by immune-effector cells such as neutrophils, macrophages and natural killer cells by inducing complement-dependent cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC). MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase. (Reproduced with permission: Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006 Sep; 6(9): 714–727). A full color version of this figure is available at Eye online.
Epidermal growth factor receptor inhibitors
There are currently five EGFR inhibitors on the market: afatinib, erlotinib, gefitinib, laptinib and vandetanib. EGFR, with its downstream effect on epidermal growth factor (EGF), is important in corneal healing and proliferation of Meibomian gland epithelial cells. In addition EGFR controls hair follicle differentiation.28 As a result, EGFR inhibitors can cause keratitis, dry eye, conjunctivitis, episcleritis, blepharitis, ectropion, entropion and eyelash trichomegaly.27, 79, 80 There are also reports of uveitis associated with EGFR kinase inhibitors.26 Rao et al81 documented a case of bilateral retinochoroiditis due to toxoplasmosis in the setting of erlotinib therapy.
Vascular endothelial growth factor receptor inhibitors
The seven VEGFR inhibitors are axitinib, lenvatinib, nintedanib, pazopanib, regorafenib, sorafenib and sunitinib. Neuro-ophthalmic toxicity seems to be infrequent with these agents as demonstrated by the few reports of neurosensory retinal detachment and PRES.25, 82, 83
Breakpoint cluster region-abelson inhibitors
As mentioned above, the first FDA-approved small-molecule kinase inhibitor was the breakpoint cluster region-abelson (Bcr-abl) inhibitor imatinib. Currently, there are four other Bcr-abl inhibitors: bosutinib, dasatinib, nilotinib and ponatinib. Thirty to 70% of patients taking imatinib develop periorbital edema.27, 28 In some cases, the periorbital edema can cause visual impairment requiring surgical intervention.84 Other complications include epiphora, keratitis, and conjunctival hemorrhage.25, 28 Retinal hemorrhage and macular edema have also been associated with imantinib.25, 85, 86
Two cases of optic neuritis in the setting of imantinib have been reported.87, 88 Details are provided for one of the cases, which involved bilateral visual loss with normal neuroimaging and cerebrospinal fluid results. Discontinuation of imantinib and institution of oral steroids was associated with an improvement of vision to ‘normal’ from counting fingers.87 Optic disc edema has been reported with imantinib.89, 90 In the case reported by Kwon et al, the patient had bilateral disc edema (with normal visual function) that resolved with discontinuation of imantinib. When the medication was restarted the disc edema did not recur. The intracranial pressure of the patient was not measured when the optic nerves were edematous.89
Mitogen-activated protein kinase kinase inhibitors
Although, Trametinib and cobimetinib are the only mitogen-activated protein kinase kinase (also known as MEK- the abbreviation of which is derived from MAPK/ERK kinase) inhibitors in clinical use, there are many molecules that are in various phases of clinical development.91 MEK is one of a series of protein kinases within the mitogen-activated protein kinase (MAPK) pathway (RAS–RAF-MEK-ERK).92 The package insert lists retinal vein occlusion (incidence 0.2%) and retinal pigment epithelial detachment as ocular toxicities associated with trametinib.93 One case report documented cystoid macular edema with trametinib.94 Draganova et al95 described a 55-year-old woman who developed bilateral panuveitis, chorioretinal folds and serous retinal detachments during trametinib and dabrafenib (see below) therapy.
Of those MEK inhibitors still being investigated in clinical trials a multitude of AEs have been identified including keratitis, epiphora, eyelid edema, blurred vision, double vision, visual disturbances (colored spots, halos), retinopathy (macular edema, central serous retinopathy, serous retinal detachment), and optic neuropathy.25, 27, 80, 94, 96, 97, 98 Intracranial hemorrhage has been reported with the use of trametinib and dabrafenib.9993
B-raf inhibitors
Dabrafenib and vemurafenib are the only two B-Raf inhibitors available on the market. Similar to MEK, B-Raf is part of the MAPK cascade. There are many similar molecules that are being investigated in clinical trials and as of yet have not received FDA approval.100, 101
The package inserts for both dabrafenib and vemurafenib list uveitis in the warning and precautions sections.102, 103 There are also reports of retinal vein occlusion occurring in patients taking B-Raf inhibitors.26
Bruton’s tyrosine kinase inhibitors
Ibrutinib is the only Bruton’s tyrosine kinase (BTK) inhibitor currently on the market. To date, there has been no ocular toxicity associated with ibrutinib. However, the package insert cautions the possibility of fatal intracranial hemorrhage.104 A fatal case of PML was described in a 75-year-old man treated with ibrutinib; however, he was also treated with rituximab and other chemotherapeutic drugs several years prior to presentation.105
Anaplastic lymphoma kinase inhibitors
Crizotinib and ceritinib are the two anaplastic lymphoma kinase (ALK) inhibitors being used in clinical practice. In two open-label, randomized, active-controlled trials reported in the package insert for crizotinib, 60–70% of patients experienced a ‘vision disorder’ defined as diplopia, photophobia, photopsia, blurred vision, reduced visual acuity, visual impairment, and vitreous floaters.106 Light to dark visual adjustment difficulties have been described by patients taking crizotinib.24, 28 In the warning and precautions section of the crizotinib package insert, the incidence of severe visual loss is estimated at 0.2% with a particular mention of optic nerve disorders.106 Chun et al reported a 69-year-old woman treated with crizotinib for metastatic lung adenocarcinoma who developed no light perception vision in the left eye and a visual field defect in the right eye. There was no mention of the appearance of the optic nerves on clinical examination but MRI demonstrated bilateral optic nerve enhancement.107 The following statement is taken directly from the package insert: ‘Discontinue XALKORI in patients with new onset of severe visual loss (best corrected vision less than 20/200 in one or both eyes). Perform an ophthalmological evaluation consisting of best corrected visual acuity, retinal photographs, visual fields, optical coherence tomography, and other evaluations as appropriate for new onset of severe visual loss. There is insufficient information to characterize the risks of resumption of XALKORI in patients.’106
Based on the crizotinib package insert approximately 19–21% of patients experience gait disturbance, hypoesthesia, muscular weakness, neuralgia, peripheral neuropathy, paresthesia, peripheral sensory neuropathy, polyneuropathy, and sensory disturbance.106
Janus kinase inhibitors
Ruxolitinib is the only Janus kinase (JAK) inhibitor on the market. No significant ocular toxicities have been associated with ruxolitinib. However, a case of bilateral retinitis due to toxoplasmosis was reported in a patient being treated with ruxolitinib.108 PML has been associated with ruxolitinib and is mentioned in the package insert.109, 110
Treatment
Depending on the severity of the AE, supportive therapy and discontinuation of the medication is often all that is needed. Infectious complications such as with toxoplasmosis can be treated with atovaquone or combination of trimethoprim-sulfamethoxazole and clindamycin.81, 108 Non-infectious uveitis can be effectively treated with topical steroids.95 Systemic corticosteroids have been used for cases of macular edema and optic neuritis.85, 87 The subretinal fluid associated with the MEK inhibitors can sometimes resolve spontaneously without interrupting therapy.111 Weber et al98 recommend continuing MEK inhibitors if subretinal fluid is present because in the majority of cases the visual symptoms are mild and transient.
Conclusions
There has been tremendous advancement in the medical treatment of cancer in the past two decades. Molecularly targeted and immune therapies hold the promise of more personalized and selective treatment. However, the introduction of these new cancer therapies with unique MOAs that carry the capability to interfere with critical intracellular signal pathways, modulate the immune system and in some cases activate the immune system, novel AEs have become a clinical challenge that physicians involved in the care of cancer patients should recognize. In addition, physicians should be prepared to institute a management plan to minimize permanent ocular and neuro-ophthalmic complications.
Method of literature search
Multiple resources were enlisted to gather the necessary the information and data for this review article. A detailed PubMed search (http://www.ncbi.nlm.nih.gov/pubmed) was performed as well as a Google search (https://www.google.com/) using the terms cancer treatment, monoclonal antibodies, immune checkpoint inhibitors, immunotherapy, chemotherapy, aromatase inhibitors, and small-molecule kinase inhibitors. Additional searches were performed depending on the initial results retrieved. Case reports, case series and published randomized clinical trials were also reviewed when necessary. Additional information on individual cancer drugs was collected from the package insert, FDA Adverse Event Reporting System (FAERS; http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/) and Medwatch (http://www.fda.gov/Safety/MedWatch/default.htm).
References
Siegel RL, Miller KD, Jemal A . Cancer Statistics, 2017. CA Cancer J Clin 2017; 67 (1): 7–30.
Gerber DE . Targeted therapies: a new generation of cancer treatments. Am Fam Physician 2008; 77 (3): 311–319.
Aggarwal S . Targeted cancer therapies. Nat Rev Drug Discov 2010; 9 (6): 427–428.
Kocher R, Roberts B . The calculus of cures. N Engl J Med 2014; 370 (16): 1473–1475.
Scharf O, Colevas AD . Adverse event reporting in publications compared with sponsor database for cancer clinical trials. J Clin Oncol 2006; 24 (24): 3933–3938.
Zhang S, Liang F, Tannock I . Use and misuse of common terminology criteria for adverse events in cancer clinical trials. BMC Cancer 2016; 16: 392.
Seruga B, Sterling L, Wang L, Tannock IF . Reporting of serious adverse drug reactions of targeted anticancer agents in pivotal phase III clinical trials. J Clin Oncol 2011; 29 (2): 174–185.
Niraula S, Seruga B, Ocana A, Shao T, Goldstein R, Tannock IF et al. The price we pay for progress: a meta-analysis of harms of newly approved anticancer drugs. J Clin Oncol 2012; 30 (24): 3012–3019.
Charlton P, Spicer J . Targeted therapy in cancer. Medicine 2016; 44 (1): 34–38.
Huang M, Shen A, Ding J, Geng M . Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci 2014; 35 (1): 41–50.
Kohler G, Milstein C . Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256 (5517): 495–497.
Liu AY, Robinson RR, Hellstrom KE, Murray ED Jr, Chang CP, Hellstrom I . Chimeric mouse-human IgG1 antibody that can mediate lysis of cancer cells. Proc Natl Acad Sci USA 1987; 84 (10): 3439–3443.
Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G . From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 2007; 25 (10): 1134–1143.
Carter P . Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001; 1 (2): 118–129.
Kontermann RE, Brinkmann U . Bispecific antibodies. Drug Discov Today 2015; 20 (7): 838–847.
Schrama D, Reisfeld RA, Becker JC . Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006; 5 (2): 147–159.
International Nonproprietary Name (INN) Working Group Meeting on Nomenclature for Monoclonal Antibodies. Available at: https://www.ama-assn.org/about/monoclonal-antibodies.
Di Martino S, Rainone A, Troise A, Di Paolo M, Pugliese S, Zappavigna S et al. Overview of FDA-approved anticancer drugs used for targeted therapy. WCRJ 2015; 3 (3): e553.
Ecker DM, Jones SD, Levine HL . The therapeutic monoclonal antibody market. MAbs 2015; 7 (1): 9–14.
Scott AM, Wolchok JD, Old LJ . Antibody therapy of cancer. Nat Rev Cancer 2012; 12 (4): 278–287.
Redman JM, Hill EM, AlDeghaither D, Weiner LM . Mechanisms of action of therapeutic antibodies for cancer. Mol Immunol 2015; 67 (2 Pt A): 28–45.
Brennan FR, Morton LD, Spindeldreher S, Kiessling A, Allenspach R, Hey A et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010; 2 (3): 233–255.
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ . The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 2010; 9 (4): 325–338.
Liu CY, Francis JH, Brodie SE, Marr B, Pulido JS, Marmor MF et al. Retinal toxicities of cancer therapy drugs: biologics, small molecule inhibitors, and chemotherapies. Retina 2014; 34 (7): 1261–1280.
Ho WL, Wong H, Yau T . The ophthalmological complications of targeted agents in cancer therapy: what do we need to know as ophthalmologists? Acta Ophthalmol 2013; 91 (7): 604–609.
Huillard O, Bakalian S, Levy C, Desjardins L, Lumbroso-Le Rouic L, Pop S et al. Ocular adverse events of molecularly targeted agents approved in solid tumours: a systematic review. Eur J Cancer 2014; 50 (3): 638–648.
Renouf DJ, Velazquez-Martin JP, Simpson R, Siu LL, Bedard PL . Ocular toxicity of targeted therapies. J Clin Oncol 2012; 30 (26): 3277–3286.
Hager T, Seitz B . Ocular side effects of biological agents in oncology: what should the clinician be aware of? Onco Targets Ther 2013; 7: 69–77.
Bosch X, Saiz A, Ramos-Casals M Group BS. Monoclonal antibody therapy-associated neurological disorders. Nat Rev Neurol 2011; 7 (3): 165–172.
Genzyme. Alemtuzumab/Campath. 2014. http://www.campath.com/pdfs/2014-09-Campath_US_PI.pdf.
Cossburn M, Pace AA, Jones J, Ali R, Ingram G, Baker K et al. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology 2011; 77 (6): 573–579.
Daniels GH, Vladic A, Brinar V, Zavalishin I, Valente W, Oyuela P et al. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab 2014; 99 (1): 80–89.
Isidoro L, Pires P, Rito L, Cordeiro G . Progressive multifocal leukoencephalopathy in a patient with chronic lymphocytic leukaemia treated with alemtuzumab. BMJ Case Rep 2014. epub ahead of print 8 January 2014 doi:10.1136/bcr-2013-201781.
Zaheer F, Berger JR . Treatment-related progressive multifocal leukoencephalopathy: current understanding and future steps. Ther Adv Drug Saf 2012; 3 (5): 227–239.
Ellis LM . Mechanisms of action of bevacizumab as a component of therapy for metastatic colorectal cancer. Semin Oncol 2006; 33 (5 Suppl 10): S1–S7.
Sherman JH, Aregawi DG, Lai A, Fathallah-Shaykh HM, Bierman PJ, Linsky K et al. Optic neuropathy in patients with glioblastoma receiving bevacizumab. Neurology 2009; 73 (22): 1924–1926.
Zuo PY, Chen XL, Liu YW, Xiao CL, Liu CY . Increased risk of cerebrovascular events in patients with cancer treated with bevacizumab: a meta-analysis. PLoS One 2014; 9 (7): e102484.
Seet RC, Rabinstein AA . Clinical features and outcomes of posterior reversible encephalopathy syndrome following bevacizumab treatment. QJM 2012; 105 (1): 69–75.
Thomas A, Teicher BA, Hassan R . Antibody-drug conjugates for cancer therapy. Lancet Oncol 2016; 17 (6): e254–e262.
White MK, Khalili K . Pathogenesis of progressive multifocal leukoencephalopathy—revisited. J Infect Dis 2011; 203 (5): 578–586.
Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 2009; 10 (8): 816–824.
Information for Healthcare Professionals: Rituximab (marketed as Rituxan). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126519.htm.
Vermeer NS, Straus SM, Mantel-Teeuwisse AK, Hidalgo-Simon A, Egberts AC, Leufkens HG et al. Drug-induced progressive multifocal leukoencephalopathy: Lessons learned from contrasting natalizumab and rituximab. Clin Pharmacol Ther 2015; 98 (5): 542–550.
Genetech. Rituxumab/Rituxan. 2012. https://www.gene.com/download/pdf/rituxan_prescribing.pdf.
Keene DL, Legare C, Taylor E, Gallivan J, Cawthorn GM, Vu D . Monoclonal antibodies and progressive multifocal leukoencephalopathy. Can J Neurol Sci 2011; 38 (4): 565–571.
Tavazzi E, Ferrante P, Khalili K . Progressive multifocal leukoencephalopathy: an unexpected complication of modern therapeutic monoclonal antibody therapies. Clin Microbiol Infect 2011; 17 (12): 1776–1780.
Genentech. Trastuzumab-Herceptin 2016. https://www.gene.com/download/pdf/herceptin_prescribing.pdf.
Saleh M, Bourcier T, Noel G, Speeg-Schatz C, Gaucher D . Bilateral macular ischemia and severe visual loss following trastuzumab therapy. Acta Oncol 2011; 50 (3): 477–478.
Landi D, De Rossi N, Zagaglia S, Scarpazza C, Prosperini L, Albanese M et al. No evidence of beneficial effects of plasmapheresis in natalizumab-associated PML. Neurology 2017; 88 (12): 1144–1152.
Molloy ES, Calabrese CM, Calabrese LH . The risk of progressive multifocal leukoencephalopathy in the biologic era: prevention and management. Rheum Dis Clin North Am 2017; 43 (1): 95–109.
Singer S, Grommes C, Reiner AS, Rosenblum MK, DeAngelis LM . Posterior reversible encephalopathy syndrome in patients with cancer. Oncologist 2015; 20 (7): 806–811.
Mellman I, Coukos G, Dranoff G . Cancer immunotherapy comes of age. Nature 2011; 480 (7378): 480–489.
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016; 54: 139–148.
Spain L, Diem S, Larkin J . Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev 2016; 44: 51–60.
Goldstein BL, Gedmintas L, Todd DJ . Drug-associated polymyalgia rheumatica/giant cell arteritis occurring in two patients after treatment with ipilimumab, an antagonist of ctla-4. Arthritis Rheumatol 2014; 66 (3): 768–769.
Johnson DB, Saranga-Perry V, Lavin PJ, Burnette WB, Clark SW, Uskavitch DR et al. Myasthenia Gravis induced by ipilimumab in patients with metastatic melanoma. J Clin Oncol 2015; 33 (33): e122–e124.
Gerdes LA, Held K, Beltran E, Berking C, Prinz JC, Junker A et al. CTLA4 as immunological checkpoint in the development of multiple sclerosis. Ann Neurol 2016; 80 (2): 294–300.
Antoun J, Titah C, Cochereau I . Ocular and orbital side-effects of checkpoint inhibitors: a review article. Curr Opin Oncol 2016; 28 (4): 288–294.
Mantopoulos D, Kendra KL, Letson AD, Cebulla CM . Bilateral choroidopathy and serous retinal detachments during ipilimumab treatment for cutaneous melanoma. JAMA Ophthalmol 2015; 133 (8): 965–967.
Yeh OL, Francis CE . Ipilimumab-associated bilateral optic neuropathy. J Neuroophthalmol 2015; 35 (2): 144–147.
Hahn L, Pepple KL . Bilateral neuroretinitis and anterior uveitis following ipilimumab treatment for metastatic melanoma. J Ophthalmic Inflamm Infect 2016; 6 (1): 14.
Sheldon CA, Kharlip J, Tamhankar MA . Inflammatory orbitopathy associated with Ipilimumab. Ophthal Plast Reconstr Surg 2015; 33 (3S Suppl 1): S155–S158.
Papavasileiou E, Prasad S, Freitag SK, Sobrin L, Lobo AM . Ipilimumab-induced ocular and orbital inflammation-A case series and review of the literature. Ocul Immunol Inflamm 2016; 24 (2): 140–146.
Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol 2016; 27 (4): 559–574.
Roberts P, Fishman GA, Joshi K, Jampol LM . Chorioretinal lesions in a case of melanoma-associated retinopathy treated with pembrolizumab. JAMA Ophthalmol 2016; 134 (10): 1184–1188.
Friedman CF, Proverbs-Singh TA, Postow MA . Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol 2016; 2 (10): 1346–1353.
Joshi MN, Whitelaw BC, Palomar MT, Wu Y, Carroll PV . Immune checkpoint inhibitor-related hypophysitis and endocrine dysfunction: clinical review. Clin Endocrinol (Oxf) 2016; 85 (3): 331–339.
Wilson MA, Guld K, Galetta S, Walsh RD, Kharlip J, Tamhankar M et al. Acute visual loss after ipilimumab treatment for metastatic melanoma. J Immunother Cancer 2016; 4: 66.
Williams TJ, Benavides DR, Patrice KA, Dalmau JO, de Avila AL, Le DT et al. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol 2016; 73 (8): 928–933.
Gonzalez NL, Puwanant A, Lu A, Marks SM, Zivkovic SA . Myasthenia triggered by immune checkpoint inhibitors: New case and literature review. Neuromuscul Disord 2017; 27 (3): 266–268.
Martin GS . Cell signaling and cancer. Cancer Cell 2003; 4 (3): 167–174.
Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP . Targeting cancer with kinase inhibitors. J Clin Invest 2015; 125 (5): 1780–1789.
Faivre S, Djelloul S, Raymond E . New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol 2006; 33 (4): 407–420.
Levitzki A, Klein S . Signal transduction therapy of cancer. Mol Aspects Med 2010; 31 (4): 287–329.
Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J et al. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther 2002; 93 (2-3): 79–98.
Rosland GV, Engelsen AS . Novel points of attack for targeted cancer therapy. Basic Clin Pharmacol Toxicol 2015; 116 (1): 9–18.
Iqbal N, Iqbal N . Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract 2014; 2014: 357027.
Wu P, Nielsen TE, Clausen MH . FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 2015; 36 (7): 422–439.
Celik T, Kosker M . Ocular side effects and trichomegaly of eyelashes induced by erlotinib: a case report and review of the literature. Cont Lens Anterior Eye 2015; 38 (1): 59–60.
Agustoni F, Platania M, Vitali M, Zilembo N, Haspinger E, Sinno V et al. Emerging toxicities in the treatment of non-small cell lung cancer: ocular disorders. Cancer Treat Rev 2014; 40 (1): 197–203.
Rao V, Schneider E, Proia AD, Fekrat S . Development of bilateral acquired toxoplasmic retinochoroiditis during erlotinib therapy. JAMA Ophthalmol 2014; 132 (9): 1150–1152.
Wegner A, Khoramnia R . Neurosensory retinal detachment due to sunitinib treatment. Eye (Lond) 2011; 25 (11): 1517–1518.
Kapiteijn E, Brand A, Kroep J, Gelderblom H . Sunitinib induced hypertension, thrombotic microangiopathy and reversible posterior leukencephalopathy syndrome. Ann Oncol 2007; 18 (10): 1745–1747.
McClelland CM, Harocopos GJ, Custer PL . Periorbital edema secondary to imatinib mesylate. Clin Ophthalmol 2010; 4: 427–431.
Georgalas I, Pavesio C, Ezra E . Bilateral cystoid macular edema in a patient with chronic myeloid leukaemia under treatment with imanitib mesylate: report of an unusual side effect. Graefes Arch Clin Exp Ophthalmol 2007; 245 (10): 1585–1586.
Gulati AP, Saif MW . Retinal neovascularization and hemorrhage associated with the use of imatinib (Gleevec((R))) in a patient being treated for gastrointestinal stromal tumor (GIST). Anticancer Res 2012; 32 (4): 1375–1377.
Govind Babu K, Attili VS, Bapsy PP, Anupama G . Imatinib-induced optic neuritis in a patient of chronic myeloid leukemia. Int Ophthalmol 2007; 27 (1): 43–44.
Breccia M, Gentilini F, Cannella L, Latagliata R, Carmosino I, Frustaci A et al. Ocular side effects in chronic myeloid leukemia patients treated with imatinib. Leuk Res 2008; 32 (7): 1022–1025.
Kwon SI, Lee DH, Kim YJ . Optic disc edema as a possible complication of Imatinib mesylate (Gleevec). Jpn J Ophthalmol 2008; 52 (4): 331–333.
DeLuca C, Shenouda-Awad N, Haskes C, Wrzesinski S . Imatinib mesylate (Gleevec) induced unilateral optic disc edema. Optom Vis Sci 2012; 89 (10): e16–e22.
Zhao Y, Adjei AA . The clinical development of MEK inhibitors. Nat Rev Clin Oncol 2014; 11 (7): 385–400.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773 (8): 1263–1284.
Novartis. Trametinib-Mekinist 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204114s001lbl.pdf.
Duncan KE, Chang LY, Patronas M . MEK inhibitors: a new class of chemotherapeutic agents with ocular toxicity. Eye (Lond) 2015; 29 (8): 1003–1012.
Draganova D, Kerger J, Caspers L, Willermain F . Severe bilateral panuveitis during melanoma treatment by Dabrafenib and Trametinib. J Ophthalmic Inflamm Infect 2015; 5: 17.
Stjepanovic N, Velazquez-Martin JP, Bedard PL . Ocular toxicities of MEK inhibitors and other targeted therapies. Ann Oncol 2016; 27 (6): 998–1005.
van Dijk EH, Duits DE, Versluis M, Luyten GP, Bergen AA, Kapiteijn EW et al. Loss of MAPK pathway activation in post-mitotic retinal cells as mechanism in mek inhibition-related retinopathy in cancer patients. Medicine (Baltimore) 2016; 95 (18): e3457.
Weber ML, Liang MC, Flaherty KT, Heier JS . Subretinal fluid associated with MEK inhibitor use in the treatment of systemic cancer. JAMA Ophthalmol 2016; 134 (8): 855–862.
Lee, le M, Feun L, Tan Y . A case of intracranial hemorrhage caused by combined dabrafenib and trametinib therapy for metastatic melanoma. Am J Case Rep 2014; 15: 441–443.
Roberts PJ, Der CJ . Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26 (22): 3291–3310.
Uehling DE, Harris PA . Recent progress on MAP kinase pathway inhibitors. Bioorg Med Chem Lett 2015; 25 (19): 4047–4056.
Novartis. Dabrafenib-Tafinlar 2016. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf.
Genentech. Vemurafenib-Zelboraf 2016. https://www.gene.com/download/pdf/zelboraf_prescribing.pdf.
Pharmacyclics. Ibrutinib-Imbruvica 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/205552s002lbl.pdf.
Lutz M, Schulze AB, Rebber E, Wiebe S, Zoubi T, Grauer OM et al. Progressive multifocal leukoencephalopathy after ibrutinib therapy for chronic lymphocytic leukemia. Cancer Res Treat 2016; 49 (2): 548–552.
Pfizer. Crizotinib-Xalkori 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf.
Chun SG, Iyengar P, Gerber DE, Hogan RN, Timmerman RD . Optic neuropathy and blindness associated with crizotinib for non-small-cell lung cancer with EML4-ALK translocation. J Clin Oncol 2015; 33 (5): e25–e26.
Goldberg RA, Reichel E, Oshry LJ . Bilateral toxoplasmosis retinitis associated with ruxolitinib. N Engl J Med 2013; 369 (7): 681–683.
Wathes R, Moule S, Milojkovic D . Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med 2013; 369 (2): 197–198.
Incyte. Ruxolitinib-Jakafi 2016. http://www.jakafi.com/pdf/prescribing-information.pdf.
Urner-Bloch U, Urner M, Stieger P, Galliker N, Winterton N, Zubel A et al. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann Oncol 2014; 25 (7): 1437–1441.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MTB: Novartis pharmaceuticals and Receptos.
AKSS: Bristol-Meyers Squibb, Celldex, Genentech, Merck, Immunocore, Reata.
Rights and permissions
About this article

Cite this article
Bhatti, M., Salama, A. Neuro-ophthalmic side effects of molecularly targeted cancer drugs. Eye 32, 287–301 (2018). https://doi.org/10.1038/eye.2017.222
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.222
This article is cited by
-
Ocular side effects of novel anti-cancer biological therapies
Scientific Reports (2021)
-
Ocular Toxicity of Targeted Anticancer Agents
Drugs (2021)
-
Markedly increased ocular side effect causing severe vision deterioration after chemotherapy using new or investigational epidermal or fibroblast growth factor receptor inhibitors
BMC Ophthalmology (2020)
-
Purified human anti-Tn and anti-T antibodies specifically recognize carcinoma tissues
Scientific Reports (2019)
