
Abstract
Purpose
The aim of this study was to determine a sequence of structural changes in acute posterior multifocal placoid pigment epitheliopathy (APMPPE) using optical coherence tomography-angiography (OCT-A) and comparing with other imaging modalities.
Patients and methods
Patients with a new diagnosis of acute-onset APMPPE referred to a regional specialist centre from October 2015 to October 2016 were included. Multimodal imaging employed on all patients from diagnosis included the following: fundus fluorescein angiography, indocyanine green angiography, fundus autofluorescence, spectral domain-OCT (SD-OCT), and OCT-A. All non-invasive imaging processes were repeated during follow-up.
Results
Ten eyes of five patients were included in the study, three males and two females, with a mean age of 26.2 years (range: 21–32) and a mean follow-up of 6.4 months (range: 2.6–13.3). All patients presented with bilateral disease and macular involving lesions. OCT-A imaging of the choriocapillaris was supportive of hypoperfusion at the site of APMPPE lesions during the acute phase of this condition with normalisation of choroidal vasculature during follow-up. Multimodal imaging consistently highlighted four sequential phases from presentation to resolution of active disease.
Conclusions
Multimodal imaging in patients with APMPPE in acute and long-term follow-up demonstrates a reversible choroidal hypoperfusion supporting the primary inciting pathology as a choriocapillaritis. The evolution shows resolution of the ischaemia through a defined sequence that results in persistent changes at the level of the retinal pigment epithelium and outer retina. OCT-A was able to detect preclinical changes and chart resolution at the level of the choriocapillaris.
Similar content being viewed by others

Introduction
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is an uncommon inflammatory condition that generally affects young healthy adults who present with transient acute central or paracentral vision loss related to multiple yellow creamy deep placoid lesions.1 Since the initial description of the disease by Gass in 1968, there has been a significant advance in our understanding of this disease.1 Debate persists as to the exact sequence of events in the aetiopathogenesis of APMPPE.2, 3, 4 The prevailing view has been that the primary insult is most likely to occur at the level of the inner choroid/choriocapillaris with retinal changes occurring secondary to this;5, 6, 7, 8, 9 however, definitive proof of this remains elusive. These discussions have also been extended to the nomenclature of the disease, with some authors advocating for a change in the current name to choroidopathy rather than epitheliopathy.10, 11
Previous studies have described the evolution of lesions in small case series using spectral domain-optical coherence tomography (SD-OCT)12, 13, 14 and fundus autofluorescence (FAF),12, 13, 15 and more recently optical coherence tomography-angiography (OCT-A);5, 9, 16, 17, 18 however, the appearance and evolution of the lesions in different imaging modalities at the same stage of the disease have not been extensively studied. The aim of this study is to describe the sequential evolution of APMPPE lesions based on data acquired over the course of patient follow-up using OCT-A and correlating this with other imaging modalities.
Materials and methods
Consecutive patients with an acute presentation of APMPPE diagnosed at the Regional Ocular Inflammatory Service at the Bristol Eye Hospital, Bristol (UK), from October 2015 to October 2016 were included in this retrospective case series. All patients had been clinically evaluated using multimodal imaging, including combined fundus fluorescein angiography (FFA) and indocyanine green angiography (ICGA), FAF (Spectralis HRA+OCT, Heidelberg Engineering, Heidelberg, Germany), SD-OCT, and OCT-A (AngioVue; Optovue Inc., Fremont, CA, USA) at presentation and during the follow-up.
APMPPE was diagnosed based on the phenotypic appearance of yellow creamy deep retinal placoid lesions with the classic pattern of early hypofluorescence and late hyperfluorescence on the FFA and persistent hypocyanescence on ICGA through all the time frames, regardless the clinical course after initial presentation. Data collected included demographics, concomitant symptoms (such as headache or viral prodrome), visual acuity (at baseline and at the end of follow-up), recurrence of lesions (for each eye), and duration of follow-up.
All patients were treated with a tapering regimen of systemic corticosteroids (1 mg/kg) and had extensive work-up to rule out other pathologies that could mimic APMPPE, including infectious diseases. This included a QuantiFERON-TB Gold test, serology for Syphilis, serum angiotensin-converting enzyme, complete blood count, and systemic inflammatory markers.
Results
Ten eyes of five patients (three males and two females) were included in the study with a mean age of 26.2 years (range: 21–32). All patients presented with bilateral ocular disease and had associated severe headache at presentation following a coryzal-like illness for ~1 week before presentation with visual symptoms. All patients presented to the uveitis clinic within 1 week of onset of visual symptoms. The symptom of headache disappeared after steroid treatment in all cases except in Patient 4 who was found to have associated central nervous system vasculitis with a small focus of high signal in the left corona radiata detected on gadolinium-enhanced magnetic resonance imaging of the brain. This patient also had an episode of erythema nodosum that developed a few days before the development of ophthalmic symptoms coupled with evidence of systemic inflammation with an elevated C-reactive protein (46 mg/l). Extensive work-up was carried out by the neurology team to exclude an underlying cause for her neuroradiological findings. This included lumbar puncture for microbiology and oligoclonal bands, all of which returned normal/negative results. Of the three other patients who had neuroimaging performed, no abnormalities were detected. Three out of five patients (four eyes) developed new fundus lesions during the follow-up, and in one of these the new lesions appeared in both eyes while on systemic treatment with prednisolone at the dose of 30 mg (Patient 3) once daily. The mean duration for which follow-up data were available was 6.4 months (range: 2.6–13.3). Table 1 summarises the demographic and clinical data for our cohort.
FFA and ICGA
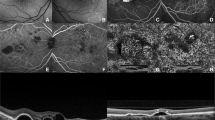
All patients had the classic FFA and ICGA findings of APMPPE at presentation as described above. Some of the lesions evident on the ICGA were not visible in the late frames of the FFA and four patients had significant peripheral choroidal disease on ICGA, which was not visible at any time point on FFA (Figures 1a–d and 2a–d).

Appearance of choroidal phase 1 lesions using different imaging modalities (left eye of Patient 3). One lesion of note is identified with a red circle. By FFA it is only evident in the early phase (a), disappearing in the late frames (b); however, it is visible on ICGA across all frames (c, d). Equally the lesion is evident on OCT-A at the level of the choriocapillaris (e1), but no changes are seen on en face-OCT (f1) or SD-OCT B-scan at that location in either the horizontal (g1, red arrow) or vertical B-scans (h1, green arrow) or fundus autofluorescence (FAF; i1). Five days from presentation the choroidal phase lesion is not seen in any of the imaging modalities, without any sequelae on FAF, en face-OCT or SD-OCT (e2–i2).

Evolution of lesions in phase 2 (right eye of Patient 3). One lesion is identified with a red circle. It is hypofluorescent in the early phase of the FFA (a) with late hyperfluorescence (b), but persistent hypocyanescence across all frames (c, d) on ICGA. OCT-A revealed an absence of flow signal (e1) at the level of the choriocapillaris that progressively improved during follow-up (e2, e3). The en face-OCT confirms that the decrease in flow at the level of the choriocapillaris is not a consequence of signal attenuation secondary to hyper-reflectivity of overlaying lesions (f). Similarly in horizontal (g) and vertical (h) sections of the SD-OCT through the lesion (red and green arrows, respectively) demonstrate that the findings evolve from an initial hyper-reflectivity at the level of the outer retina (g1, h1) to a progressive retinal thinning with disruption of the RPE layer (g2, 3 and h2, 3). On FAF (i) lesions evolved from predominant hypoautofluorescence (i1) at baseline to central hyperautofluorescence (i2), but this subsequently gave way to a recurrence of hypoautofluorescence (i3). A full color version of this figure is available at the Eye journal online.
SD-OCT
In all cases, abnormalities in the SD-OCT were detected at some stage during lesion evolution. Where patients were imaged early in the course of disease the SD-OCT may appear qualitatively normal in regions that subsequently become abnormal (Figure 3). The earliest abnormalities detected were subtle irregularities in the inner segment ellipsoid (ISe) layer and retinal pigment epithelium (RPE) with associated loss of the hyporeflective space normally visualised between these layers on SD-OCT. The latter change tended to be observed primarily in the central macula where this space is more readily identifiable in healthy controls. As lesions became more established and/or larger, focal areas of hyper-reflectivity also developed in the outer retina, primarily localising to the outer nuclear layer (ONL) but also being seen at the level of the outer plexiform layer. An intra-retinal cyst was noted at the level of the ONL in one eye. SD-OCT also revealed areas of possible focal subretinal fluid in association with larger APMPPE lesions at baseline. In lesions that had become established and failed to completely resolve, there was evidence of persistent disruption and/or irregularity of the RPE and associated retinal thinning during follow-up. In other, usually smaller lesions, improvement and almost complete resolution of the detectable abnormalities in the ISe and external limiting membrane (ELM) were observed (Figure 2g1).

Evolution of Phase 1 lesion to a Phase 2 lesion and consecutive stages (left eye of Patient 3). The lesion is identified with a red circle. OCT-A (a1) discloses a small area of hypoperfusion, equally en face-OCT shows mild hyporeflectivity at the level of the choriocapillaris (b1), but FAF and SD-OCT are essentially normal in the early stage (c1–e1). This choriocapillaris hypoperfusion identified on OCT-A had increased in size 10 days later (a2) also visible in the en face-OCT (b2) with the lesion now identifiable on the SD-OCT in the horizontal (c2, red line) and vertical B-scans (d2, green line), as well as with FAF (e2). A full color version of this figure is available at the Eye journal online.
Fundus autofluorescence
The pattern of FAF change was heterogeneous, particularly early in the course of lesion evolution. Hypoautofluorescent lesions were detected in the active stage of the disease in all patients. These often had an area of relative hyperautofluorescence along their edge. Over time there was a progressive increase in the autofluorescence signal leading to predominantly hyperautofluorescence in the centre of the lesion, but this became more generalised and extended to the periphery of the lesion during follow-up. With time this hyperautofluorescent signal became less uniform and the lesion acquired a speckled appearance. This ultimately led to hypoautofluorescence (Figures 2 and 3) consistent with localised atrophy of the RPE centrally (corresponding to fundal findings) but often hyperautofluorescence was maintained at the edge of the lesion. Individual FAF lesion area may be larger at follow-up than the area of lesions identified on FFA/ICG at baseline.
OCT-A
There was evidence of significant hypoperfusion at the level of the choriocapillaris in all active lesions of all patients at baseline. Progressive reperfusion of the choriocapillaris led to marked improvement, including complete resolution, of hypoperfusion in many lesions as detected on choroidal OCT-A (Figures 2 and 3). In three cases (see below) OCT-A abnormalities developed after normalisation following the initial onset of APMPPE. Of note, in all cases OCT-A revealed an area of relative hyper-reflectivity along the outer border of the region of choriocapillaris hypoperfusion. Furthermore, comparing the SD-OCT B-scan with the en face-OCT-A image it appears that the area of hypoperfusion extends beyond the limit of the abnormalities identified on the SD-OCT B-scan image at the level of the outer retina (Figure 4). Longitudinal follow-up of lesions revealed that both the outer retinal layers and the choriocapillaris normalise centripetally from the outer edge of the APMPPE lesion, but that the ‘leading edge’ of recovery of the retinal layers appears to precede that of the choriocapilliaris throughout follow-up. New lesions in the OCT-A were visible in two of the patients (Patient 2 and Patient 3) that relapsed without new symptoms. These were evident on FAF and SD-OCT also. In another patient (Patient 1) an OCT-A lesion actually occurred months after the initial presentation and interestingly this lesion developed in the same area as a previous subclinical lesion that had been visualised only on OCT-A.

Choriocapillaris involvement exceeds detectable retinal structural changes (right eye of Patient 5). OCT-A image (a) at the level of the choriocapillaris (parallel horizontal red lines in b) revealed focal areas of hypoperfusion in the right eye of Patient 5. Corresponding SD-OCT scan (b) at the level of the green line on the OCT-A image (a1) revealed associated abnormalities in the outer retinal layers and RPE. The outer border of the area of hypoperfusion on OCT-A was seen to extend to a greater extent than the OCT B-scan abnormalities (corresponding locations in a and b images are identified by vertical red lines). In the early evolution of the lesions the area of hypoperfusion in the OCT-A increased in size (a2), whereas the SD-OCT findings reveal qualitative outer retinal thinning (b2). With time the area of hypoperfusion at the level of the choriocapillaris improves (a3, 4) and the thinning of the outer retina in SD-OCT persists (b3, 4). Corresponding en face-OCT at the level of the choriocapillaris at each time point is presented in the bottom row (c1–4). A full color version of this figure is available at the Eye journal online.
Proposed phases of APMPPE lesions by multimodal imaging
On the basis of the characteristics of each imaging modality at each stage in the evolution of the lesion, we were able to define four distinct consecutive phases:
-
Phase 1—Choroidal: choriocapillaris hypoperfusion detected with OCT-A and confirmed with ICGA and an early FFA phenotype. Both SD-OCT and FAF are normal. These lesions can evolve resulting in complete resolution of the choriocapillaris abnormalities as detected with OCT-A and without any structural changes developing on FAF or SD-OCT (Figure 1), or they evolve to phase 2 lesions (see below; Figure 3).
-
Phase 2—Chorioretinal: evidence of classic active lesion on FFA with early hypofluorescence and late hyperfluorescence, persistent hypocyanescence of lesions across ICGA frames and choroidal hypoperfusion on OCT-A. Loss of structural integrity and hyper-reflectivity of the outer retinal layers including RPE, ISe layer, and ELM were detected on SD-OCT and predominant hypoautofluorescent of APMPPE lesions noted on FAF (Figure 2).
-
Phase 3—Transitional: thinning and disruption of outer retinal layers revealed on SD-OCT with persistent hypoperfusion of the choriocapillaris observed on OCT-A, and progressive central hyperautofluorescence observed on FAF (Figure 2).
-
Phase 4—Resolution: persistent thinning of the outer retina visible on SD-OCT with hyporeflectivity at the level of the RPE and predominantly hypoautofluorescent changes visible on FAF, but with a normalised choriocapillaris vascular pattern on OCT-A (Figure 2).
Discussion
Multimodal imaging provides valuable information and enhanced understanding of the progression of APMPPE through to clinical resolution. In our series there was a pattern of evolution of the APMPPE lesions with a clear sequence of structural changes from presentation to resolution employing multimodal imaging. The combination of OCT-A, SD-OCT, and FAF imaging has the advantage of providing clinical information without the need to use invasive injection of contrast dyes. As such, these investigations are more readily repeatable during the course of follow-up of APMPPE. Additional work is certainly required to validate the use of OCT-A in APMPPE if this is to replace conventional angiography in diagnosis and management.16
Our findings support the possibility that choriocapillaris hypoperfusion is the primary event in the pathogenesis of APMPPE, with RPE and outer retinal pathology developing secondary to the localised hypoperfusion resulting from this inner choroidal change. This is supported by the fact that lesions in the choroidal phase can resolve without leaving structural changes at the level of RPE and the area of hypoperfusion at the level of the choriocapillaris appears larger than the area of outer retina disruption on the SD-OCT, as it is clearly showed in Figure 4. A similar finding has recently been reported by Heiferman et al.18 In fact, these findings also support that the presumed hypoperfusion in the OCT-A at the level of the choriocapillaris is not due to shadowing from the hyper-reflectivity of the overlying retina. The hyper-reflectivity identified on OCT-A along the border of the area of hypoperfusion within the choriocapillaris may also represent increased blood flow along the lesion edge, and this phenotype of ‘reactive hyperperfusion’ has previously been described in Central Serous Chorioretinopathy.19
It has been proposed that APMPPE is an autolimited monophasic disease that can resolve spontaneously without any treatment,1 and the role of systemic treatment is not clear in this disease. The fact that lesions in phase 1 can resolve without any morphological sequelae could provide the opportunity to assess the role of treatment vs observation in APMPPE using features identified on multimodal imaging as end points in future prospective studies. Importantly, there is growing evidence that without treatment residual symptoms, such as paracentral scotomas, may persist.20 Equally some of the bigger lesions showed an abnormal morphology of the choriocapillaris voids in the reperfused area in the OCT-A at the last follow-up available. This has also been described by others,18 and it could be the result of the different nature of the lesions or just a question of requiring longer follow-up to detect complete resolution in larger lesions.
The sequence of events with SD-OCT,12, 13, 14 FAF,12, 13, 15 and OCT-A5, 7, 9, 16, 17, 18 has been previously described, but the correlation between different imaging technologies at the same time point has not been reported for the three imaging modalities. A previous study correlating FAF and SD-OCT findings found similar results to our study, with an initial hypoautofluorescence corresponding to the hyper-reflectance of the outer retinal layers on SD-OCT, probably due to a masking effect, with an increase in the hyperautofluorescence later when the OCT findings progress to disruption of the RPE layer.12
The authors acknowledge some important limitations in this study. Because of the retrospective nature of the study, patients did not have a standardised follow-up regime. Structure–function correlation would be interesting, particularly to determine whether resolved lesions have any residual retinal dysfunction detectable. Clearly, larger cohorts of patients would be more beneficial, particularly for the purposes of quantitative analysis/correlation.
In summary, the use of multimodal imaging for the follow-up of patients with APMPPE supports the increasing evidence of a choriocapillaritis as the underlying pathology in APMPPE, which may occur secondary to different aetiologies. Our study of the evolution of the disease shows resolution of choroidal ischaemia and an associated well-defined sequence of imaging-related descriptors that can result in either permanent changes at the level of the RPE or resolution. The grading proposal we present has the potential to provide an additional clinical tool to describe disease progression longitudinally, as well as permit the categorical assessment of effect of future therapeutic interventions for the treatment of APMPPE; however, given the small numbers on this series and the retrospective nature of this study, further prospective multicentre studies are warranted for validation.

References
Gass JD . Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1968; 80 (2): 177–185.
Bouchenaki N, Cimino L, Auer C, Tao Tran V, Herbort CP . Assessment and classification of choroidal vasculitis in posterior uveitis using indocyanine green angiography. Klin Monbl Augenheilkd 2002; 219 (4): 243–249.
Mrejen S, Gallego-Pinazo R, Wald KJ, Freund KB . Acute posterior multifocal placoid pigment epitheliopathy as a choroidopathy: what we learned from adaptive optics imaging. JAMA Ophthalmol 2013; 131 (10): 1363–1364.
Deutman AF, Boen-Tan TN, Oosterhuis JA . Proceedings: acute posterior multifocal placoid pigment epitheliopathy. Ophthalmologica 1973; 167 (5): 368–372.
Dolz-Marco R, Sarraf D, Giovinazzo V, Freund KB . Optical coherence tomography angiography shows inner choroidal ischemia in acute posterior multifocal placoid pigment epitheliopathy. Retin Cases Brief Rep 2016; 11 (Suppl 1): S136–S143.
Invernizzi A, Agarwal A, Cozzi M, Viola F, Nguyen QD, Staurenghi G . Enhanced depth imaging optical coherence tomography features in areas of choriocapillaris hypoperfusion. Retina 2016; 36 (10): 2013–2021.
Kinouchi R, Nishikawa N, Ishibazawa A, Yoshida A . Vascular rarefaction at the choriocapillaris in acute posterior multifocal placoid pigment epitheliopathy viewed on OCT angiography. Int Ophthalmol 2016; 37 (3): 733–736.
Mrejen S, Sarraf D, Chexal S, Wald K, Freund KB . Choroidal involvement in acute posterior multifocal placoid pigment epitheliopathy. Ophthalmic Surg Lasers Imaging Retina 2016; 47 (1): 20–26.
Salvatore S, Steeples LR, Ross AH, Bailey C, Lee RW, Carreno E . Multimodal imaging in acute posterior multifocal placoid pigment epitheliopathy demonstrating obstruction of the choriocapillaris. Ophthalmic Surg Lasers Imaging Retina 2016; 47 (7): 677–681.
Zhang AY, Han IC, Goldberg MF . Renaming of acute posterior multifocal placoid pigment epitheliopathy (apmppe) to acute multifocal placoid choroidopathy (AMP-C). JAMA Ophthalmol 2017; 135 (3): 185.
Jampol LM, Goldstein DA, Fawzi AA . Keeping the name of acute posterior multifocal placoid pigment epitheliopathy. JAMA Ophthalmol 2017; 135 (3): 186.
Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C . Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol 2006; 244 (10): 1219–1223.
Batioglu F, Ozmert E, Kurt R . Fundus autofluorescence and spectral optical coherence tomography findings in a case with acute posterior multifocal placoid pigment epitheliopathy. Ann Ophthalmol 2008; 40 (3-4): 185–189.
Scarinci F, Fawzi AA, Shaarawy A, Simonett JM, Jampol LM . Longitudinal quantitative evaluation of outer retinal lesions in acute posterior multifocal placoid pigment epitheliopathy using optical coherence tomography. Retina 2017; 37 (5): 851–857.
Lee CS, Lee AY, Forooghian F, Bergstrom CS, Yan J, Yeh S . Fundus autofluorescence features in the inflammatory maculopathies. Clin Ophthalmol 2014; 8: 2001–2012.
Klufas MA, Phasukkijwatana N, Iafe NA, Prasad PS, Agarwal A, Gupta V et al. Optical coherence tomography angiography reveals chorio capillaris flow reductions in placoid chorioretinitis. Ophthalmol Retina 2017; 124 (1): 2–6.
Park SS, Thinda S, Kim DY, Zawadzki RJ, Werner JS . Phase-variance optical coherence tomographic angiography imaging of choroidal perfusion changes associated with acute posterior multifocal placoid pigment epitheliopathy. JAMA Ophthalmol 2016; 134 (8): 943–945.
Heiferman MJ, Rahmani S, Jampol LM, Nesper PL, Skondra D, Kim LA et al. Acute posterior multifocal placoid pigment epitheliopathy on Optical Coherence Tomography Angiography. Retina 2017. e-pub ahead of print 1 February 2017 doi:10.1097/IAE.0000000000001487.
Teussink MM, Breukink MB, van Grinsven MJ, Hoyng CB, Klevering BJ, Boon CJ et al. OCT angiography compared to fluorescein and indocyanine green angiography in chronic central serous chorioretinopathy. Invest Ophthalmol Vis Sci 2015; 56 (9): 5229–5237.
Wolf MD, Alward WL, Folk JC . Long-term visual function in acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1991; 109 (6): 800–803.
Acknowledgements
The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
Disclaimer
The views expressed are those of the authors and are not necessarily those of the NHS, the NIHR, or the Department of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Burke, T., Chu, C., Salvatore, S. et al. Application of OCT-angiography to characterise the evolution of chorioretinal lesions in acute posterior multifocal placoid pigment epitheliopathy. Eye 31, 1399–1408 (2017). https://doi.org/10.1038/eye.2017.180
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.180
This article is cited by
-
Vessel density on optical coherence tomography angiography is prognostic for future disease course in intermediate uveitis
Scientific Reports (2024)
-
Use of optical coherence tomography angiography in the uveitis clinic
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)
-
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE)
Journal of Ophthalmic Inflammation and Infection (2021)
-
Acute posterior multifocal placoid pigment epitheliopathy after alemtuzumab treatment for relapsing–remitting multiple sclerosis
Journal of Neurology (2019)
