
Abstract
Purpose
Jalili syndrome is an autosomal recessive disorder characterized by simultaneous appearance of cone-rod dystrophy (CRD) and amelogenesis imperfecta (AI). Mutations in CNNM4 gene have been identified as the underlying cause of the syndrome. In this study, we investigated a large affected family to identify the causative mutation.
Patients and Methods
A seven-generation family with 24 members affected with Jalili syndrome were enrolled in the study. Comprehensive ophthalmologic and dental examinations were performed on them. The entire coding region of CNNM4 gene was sequenced for detection of potential mutations.
Results
Ocular examinations showed nystagmus and photophobia along with early onset visual impairment. Fundoscopic exams revealed a spectrum of macular dystrophies in different family members, from macular coloboma and advanced form of beaten bronze macular dystrophy (bull’s eye) to milder form of macular thinning along with a range of pigmentary changes and vascular attenuation in the posterior pole and periphery. Scotopic and photopic electro-retinographic responses (ERGs) were extinguished or significantly depressed. Mutation analysis revealed a novel mutation (c.1091delG) in homozygous form in the patients and as a heterozygous form in the normal carrier subjects.
Conclusion
We identified a novel homozygous deleterious mutation in CNNM4 gene which causes Jalili syndrome.
Similar content being viewed by others


Introduction
In 1988, a rare autosomal recessive oculo-dental syndrome of combined cone-rod dystrophy (CRD) and amelogenesis imperfect (AI), named Jalili syndrome (OMIM 217080), was described in three large families from the Gaza Strip.1 The first and most prevalent clinical feature of Jalili syndrome observed in most cases is ocular.2 CRD is a clinically and genetically heterogeneous retinal disorder that usually manifests in childhood. CRD initially begins with a progressive and degenerative process, which affects the cone photoreceptors. Degeneration of the rod photoreceptors typically occurs sequentially or may occur simultaneously with cone degeneration.3 This group of disorders is distinguished from rod-cone dystrophy, in which rod photoreceptors are affected earlier than cones. The term retinitis pigmentosa is now applied for the latter disorder.4 The main clinical feature of CRD is cone photoreceptor dysfunction including a progressive loss of vision, photo-aversion (photophobia), achromatopsia, nystagmus, and visual impairment followed by night blindness reflecting rod photoreceptor involvement.3, 4 Patients with CRD suffer from progressive loss of central vision that eventually ends in loss of peripheral visual fields and total blindness.
CRD occurs in both non-syndromic and syndromic forms. Thus far, several mutations in various genes have been identified which are involved in non-syndromic CRD. These genes encode a wide range of proteins, including the visual cycle components, structural components of photoreceptors, photoreceptor specific transcription factors, and the proteins involved in the neuronal development.5, 6 Syndromic CRD has been increasingly reported such as Bardet-Biedl, Rogers syndrome, or spinocerebellar ataxia type.7, 8
Amelogenesis imperfecta (AI) is another heterogeneous group of dental development disorders. AI is characterized by atypical structure and clinical appearance of the tooth enamel, which is seen in both primary and secondary dentitions and eventually ends in loss of teeth.9 Similar to CRD, AI can be inherited in isolation or as a part of a syndrome. Several mutations were found in multiple loci in AI patients, therefore, all of these genes are involved in the process of highly mineralized dental enamel formation.10 AI is also reported as a part of some other syndromes such as enamel renal syndrome, Kohlschutter-Tonz syndrome, and trichodento-osseous syndrome.6
Jalili syndrome is a disorder with wide phenotypic variability within and between the affected families.11, 12, 13 Comprehensive investigations in affected patients have revealed the putative gene at 2q11 locus. After sequencing, mutations were identified in the cyclin M4 (CNNM4) gene, thus this gene was introduced as the underlying cause of the disorder.7 The protein encoded by CNNM4 is a member of the ancient conserved domain protein family and is possibly involved in the metal ion transport, particularly magnesium, which is necessary for accurate function of the photoreceptors, and homeostasis in the retina.14, 15 CNNM4 expression has been demonstrated in the enamel and dentin-forming cells, suggesting that disturbance of the Mg2+ transport leads to mineral deficiency observed in dental enamel of AI patients.5, 7 Various kinds of mutations including missense, nonsense, deletion, and single base duplication have been identified in CNNM4 in Jalili syndrome. These mutations often take place within the conserved domains of the protein and results in loss-of-function.16
In this study, we ascertained a large consanguineous pedigree with Jalili syndrome and described the detailed ocular and dental phenotype. Moreover, we screened CNNM4 in the patients in order to detect possible causative mutations.
Subjects and methods
Patients and clinical investigations
A large consanguineous Iranian family with 24 members with diagnosis of Jalili syndrome from Chalandar village with a population of 1380 people in Mazandaran province, Iran, was included in our study (Figure 1). Considering that all families in the pedigree were inhabitants of the remote rural areas, most of them were out of access for detailed clinical investigations, thus only we could have accessed four affected members (V8, V9, V20, and IV22) for detailed clinical investigations. These patients were belonged to four different sibships and were 25 years old male (V8), 32 years old female (V9), 27 years old male (V20), and 39 years old female (IV22). Selected patients were referred to ophthalmologist and dentist for comprehensive clinical examinations. Before starting the clinical and genetic testing, informed consent was obtained from all the participants. This study was approved by the local ethics committee at Shahid Beheshti University of Medical Sciences.

The pedigree of studied Jalili syndrome family. The siblings in each family are in sequence of birth ranks.
Ocular examinations
Complete ocular examinations were performed by two academic experienced ophthalmologists. First uncorrected visual acuity was performed using the standard 20 feet Snellen acuity chart. Then external eye exams including eye movements and reverse afferent pupillary defect test were done. Best corrected visual acuity tests were obtained after manifesting refraction without eye drop. Cyclo-refraction was done after 20 min of instilling one drop of tropicamide 1% (Sina Daru, Tehran, Iran), three times for every 5 min, using manual retinoscope. Intraocular pressure was measured by Goldmann applanation tonometry test after applying one drop of ophthalmic drop tetracaine hydrochloride 0.5% (Sina Daru, Iran). Complete slit-lamp bio-microscopy examinations including cornea, anterior chamber clarity and angle, iris, pupil, lens and anterior vitreous exams were performed. Also complete macular and fundus exam including disc color, margin, cupping and vessels were performed using +90 lens and slit-lamp bio-microscopy. Then peripheral retinal exam was done using indirect ophthalmoscopy and +20 lens. Ocular coherence tomography, infra-red fundus photography, blue-light fundus auto-fluorescent and fluorescent angiography was done through Spectralis, multi-modality diagnostic imaging (Heidelberg, Germany).
Electrodiagnostic tests
Color vision test was performed using standard Ishsihara plates. Full-field flash scotopic rod responses to low intensity (0.01 cd.s/m2) blue light in dark-adapted state, scotopic maximal combined rod and cone responses to bright light flash (3 cd.s/m2) on a dark background, photopic cone responses to bright light flash (3 cd.s/m2) after light adaptation for 10 min, and 30 Hz flicker cone response electro-retinograms (ERGs) and electro-oculograms (EOGs) were performed according to the protocols recommended by ISCEV using Vision Monitor, Win8000A (Metrovision, Lille, France).
Mutation analysis
DNA samples of a core family including IV.28, IV.29, V.18, V.19, V.20, and V.21 were first extracted from peripheral blood using a standard procedure.17 PCR procedure performed for all seven exons and boundary regions of CNNM4 gene. After purification of all PCR products, sequencing was performed in both the forward and reverse directions for all exons with Applied Biosystems BigDye terminator v3.1 sequencing chemistry as per the manufacturer’s instructions on a genetic analyzer (ABI3130, Applied Biosystems, Foster City, CA, USA) and then the results were analyzed using Sequencher 5.0 software (Gene Codes Corporation, Ann Arbor, MI, USA). Considering that blood samples were available for all affected members, we also sequenced the DNA of other affected pedigree members from different sibships for the region of discovered mutation to confirm the causative mutation.
Results
Clinical manifestations
Ocular
Two of patients (IV.22 and V.20) presented with severe visual impairments along with photophobia and nystagmus since birth. V.9 had photophobia and nystagmus from early infancy with progressive visual impairment detected as moderate visual loss in early childhood progressed to severe visual loss in late adolescence. Best corrected visual acuity (BCVA) of V9 was counting finger at the time of our clinical exam. V.8 showed the mildest phenotype of this affected family. He had photophobia, latent nystagmus, and mild visual impairment since early childhood and underwent strabismus surgery at the age 11 years old.
Refractive errors in these four patients ranged from mild to moderate hyperopia and astigmatism (Table 1). BCVA of each patient is also presented in Table 1. Their vision was compromized under outdoor illumination and partially improved under indoor conditions. Generally, they did not complain of night blindness, except V.8, who reported reduced vision under very low illuminations.
The eye movement abnormality in IV.22 and V.20 was almost the same as wandering eyes. In V.9 nystagmus was in the form of fine pendular that aggravated in the daylight outdoor illumination. V.8 nystagmus was of latent pendular type, which manifested under daylight outdoor illumination.
Anterior segment examinations showed clear cornea, deep, formed and clear anterior chamber, with round and mobile pupil. Mild posterior sub-capsular opacity was seen in V8. Anterior polar opacity and blue-dot cataract was noted in IV.22 and V20, but V.9 had clear lens.
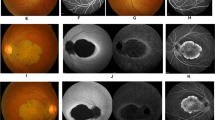
Fundus examinations using slit-lamp biomicroscopy and +90 lens, revealed an area of no neuro-retina and no retinal pigmented epithelium of about three disc areas diameter in the macula with large choroidal vessels passing beneath it, referring as macular coloboma, symmetrically in both the eyes of V.20 and IV.22 patients (Figure 2c). The size of coloboma was larger in V.20, which involved almost the entire macula. Advanced macular atrophy with Bull’s eye appearance was noticed in V.9 (Figure 2a). Optical coherence tomography (OCT) was not possible to achieve in any of the above mentioned patients except V.8, which showed mild diffuse macular thinning and reduced total macular volume. Pigment clumps were seen around the macular coloboma in V.20 and IV.22, in addition to diffusely scattered pigments in the retinal mid-periphery and along the retinal vessels (Figure 2c). Fine pigment clumping was scattered in the retinal mid and far periphery and around the retinal vessels in V.8 and V.9 eyes. In addition, whitish deep dots were seen through the entire posterior pole and in the retinal mid and far periphery in V.9 fundus examination (Figure 2a). The area with loss of fundus auto-fluorescent (FAF) imaging in V.9 was compatible with the area of macular retinal pigment epithelial (RPE) atrophy and loss of macular lipofuscin granules (Figure 2b). Lack of any detectable auto-fluorescence in FAF image of IV.22 patient was also compatible with large area of macular coloboma and diffuse RPE atrophy around the macula and along the entire posterior pole (Figure 2d). It was not possible to take color fundus photograph and FAF from V.20 patient because of lack of any fixation, severe nystagmus, and photophobia. Different severities of disc pallor was observed in patients as the waxy pallor discs in IV.22 and V20, moderate disc pallor and optic atrophy in V9 and mild optic atrophy in V8. Attenuation of retinal vessels were observed in all these four patients, which was more pronounced in the eldest person, IV.22 (Figure 2a, c and e).

Color fundus photograph and fundus auto-fluorescent (FAF) images. (a) Color fundus photograph shows advanced macular atrophy (beaten bronze) along with white pigment clumps in mid-periphery and around the retinal vessels; (b) FAF shows loss of macular auto-fluorescent due to retinal pigment epithelium and photoreceptor atrophy in macula in V.9. (c) Color fundus photograph shows macular coloboma with pigment clumps around coloboma in IV.22 and (d) Total loss of fundus auto-fluorescent in IV.22. (e) Color fundus photograph depicts nearly normal macula with decreased foveal reflex and pigment clumps in mid-periphery and along attenuated vessels in V.8. (f) Shows nearly normal auto-fluorescence of macula along with lower amounts of auto-fluorescence of mid-periphery of retina (outside vascular arcades). Different degrees of disc pallor and vessel attenuation are also visible in fundus photography of V.9, IV.22, and V.8 patients (a, c, and e).
OCT in V.8, showed diffuse macular atrophy with preserved foveal contour in both the eyes.
Electrodiagnostic test results
Color blindness was detected in all the four patients. Full field scotopic rod flash ERG responses to low-intensity blue light in dark adapted state were flat in all patients. Amplitudes of scotopic rod and cone maximal combined responses to high-intensity white light with dark background were nearly extinguished in IV.22, V.20 and V.9, and severely depressed in V.8 in comparison to normal control (Figure 3). Amplitudes of photopic cone responses to bright white light flash under light adapted conditions and 30 Hz flicker responses were severely reduced in V.8, but were extinguished in V.9, V.20, and IV.22.

Full-field scotopic maximal combined rod and cone ERG responses to white light flash (3 cd.s/m2 light intensity). (a) Normal amplitudes of a and b wave responses. (b and c) Extinguished or flat ERG (no detectable a and b waves) in IV.22, V.20 patients. (d) Nearly flat b-wave with no detectable a-wave in V9. (e) Severely depressed a-wave and b-wave amplitudes inV8.
EOG was undetectable in IV.22 and V.20 eyes. But subnormal Arden ratios were detected in V9 and V8.
Dental
Four patients underwent a detailed dental examination. All examined patients were diagnosed with AI.
Mutation analysis
DNA sequencing analysis for CNNM4 gene revealed a novel mutation in homozygote state in the affected individuals of the pedigree. The mutation was present in heterozygous state in the parents of all affected individuals. The mutation was a G nucleotide deletion (c.1091delG) in the first exon of the CNNM4 gene, resulting in a frameshift which eventually ends in a premature stop codon after six codons downstream the mutation position (p.G364Vfs*9) (Figure 4).

The site of causative mutation in normal, carrier, and patients with Jalili syndrome.
Discussion
Here, we described a large family with the autosomal recessive Jalili syndrome. The mode of inheritance was concluded from the high rate of consanguinity in the pedigree. In two out of our four investigated patients, the retinal dystrophy was of early neonatal onset with severe visual impairment. In one patient retinal dystrophy manifested as early infantile type but with severely progressive pattern. Although retinal dystrophy in one patient was of milder form which started from childhood and progressed with less severity as in the age 25 years old had at least Snellen chart vision. All four patients had nystagmus, photophobia, and optic atrophy from mild pattern to severe or waxy pallor disc.
In the first reported family with 29 affected members across seven generations described by Jalili, night blindness was not observed and scotopic ERGs were only slightly reduced in the 12-years-old subject.1 In another investigation performed by Michaelides et al4 retinal findings showed similar results as Arabic report but two brothers in their family reported early onset night blindness, which their rod ERGs were profoundly abnormal. Accordingly, findings from our family revealed extinguished to severely depressed ERG responses in both scotopic rod and maximal combined rod and cone responses along with photopic and 30 Hz flicker cone responses, which denotes involvement of both cone and rod photoreceptors. One patient with less severe form of retinal dystrophy also reported night blindness.
AI divides in two hypoplastic and hypomineralized forms based on the histological and morphological alterations in enamel, but there may be overlap between these types.9 In Arabian study, the type of AI was hypomineralized.1 and it was hypoplastic/hypomineralized type in Kosovan family.4
CNNM4 protein is belonged to the cyclin mediator (CNNM) family proteins, defined to be conserved Mg2+ transporters. Mutations in the CNNM4 show clinical consequences, limited to retinal function and bio-mineralization of teeth, with CRD and AI phenotypic manifestations.5 Deletion mutants of CNNM4, lacking the CBS (cystathionine-beta-synthase) domains, are unable to promote Mg2+ efflux (16).
The mutation found in our patients results in a premature stop codon that may undergo nonsense mediated decay, a conserved cellular surveillance pathway, which degrades mRNAs harboring premature stop codons, in order to prevent the production of a nonfunctional or even harmful products.18 However, it is also likely that the mRNA translates to a short truncated protein.
The mutation we have identified occurred in the first exon of the gene, which can possibly result in a protein lacking of almost all critical domains including CSB domains. Thus, the ability of the protein to regulate the transport of Mg2+, encounters with failure. However, the functional consequences of the mutation are yet to be established in the subsequent researches. To the best of our knowledge, 15 different mutations have been detected in the CNNM4 gene around the world and different mutations show variable interfamilial and intrafamilial expression.6, 19, 20, 21, 22 Types of mutations occurring in Jalili syndrome address a clinical heterogeneity and suggest loss of function as the underlying mechanism.7 Here, we report variable expressions of cone-rod retinal dystrophy in terms of age of onset, severity in visual impairment and progression, in four patients of our family. Our study presented a comprehensive clinical picture of the rare autosomal recessive Jalili syndrome in a large consanguineous family and found a new homozygous deleterious mutation in patients, which existed in carriers in heterozygote form.

References
Jalili IK, Smith NJ . A progressive cone-rod dystrophy and amelogenesis imperfecta: a new syndrome. J Med Genet 1988; 25 (11): 738–740.
Doucette L, Green J, Black C, Schwartzentruber J, Johnson GJ, Galutira D et al. Molecular genetics of achromatopsia in newfoundland reveal genetic heterogeneity, founder effects and the first cases of Jalili syndrome in North America. Ophthalmic Genet 2013; 34 (3): 119–129.
Hamel CP . Cone rod dystrophies. Orphanet J Rare Dis 2007; 2: 7.
Michaelides M, Bloch-Zupan A, Holder GE, Hunt DM, Moore AT . An autosomal recessive cone-rod dystrophy associated with amelogenesis imperfecta. J Med Genet 2004; 41 (6): 468–473.
Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet 2009; 84 (2): 266–273.
Nair P, Obeid T, Tadmouri G, Al-Khaja N, Jalili IK . A study in pleiotropy Jalili syndrome. Hamdan Med J 2013; 6: 233–240.
Polok B, Escher P, Ambresin A, Chouery E, Bolay S, Meunier I et al. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am J Hum Genet 2009; 84 (2): 259–265.
Jalili IK . Retinal ciliopathies: list of cone-rod ciliopathies and mutations in Jalili syndrome http://jalili.co/crc/crc_t2011.pdf. (accessed 25 February 2013).
Aldred MJ, Savarirayan R, Crawford PJ . Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral Dis 2003; 9 (1): 19–23.
Crawford PJ, Aldred M, Bloch-Zupan A . Amelogenesis imperfecta. Orphanet J Rare Dis 2007; 2: 17.
Jalili IK . Cone-rod dystrophy and amelogenesis imperfecta (Jalili Syndrome): phenotypes and environs. Eye 2010; 24 (11): 1659–1668.
Gerth-Kahlert C, Seebauer B, Dold S, Hanson JV, Wildberger H, Sporri A et al. Intra-familial phenotype variability in patients with Jalili syndrome. Eye 2015; 29 (5): 712–716.
Purwar P, Sareen S, Bhartiya K, Sayed Inayatullah SR, Bansal M, Chahal V et al. Jalili syndrome presenting with situs inversus totalis and keratoconus: the first case in the indian subcontinent. Oral Surg Oral Med Oral Pathol Oral Radiol 2015; 120 (5): e210–e218.
Goytain A, Quamme GA . Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics 2005; 22 (3): 382–389.
Meyer TE, Verwoert GC, Hwang SJ, Glazer NL, Smith AV, van Rooij FJ et al. Genome-wide association studies of serum magnesium, potassium, and sodium concentrations identify six loci influencing serum magnesium levels. PLoS Genet 2010; 6 (8): pii: e1001045.
Luder HU, Gerth-Kahlert C, Ostertag-Benzinger S, Schorderet DF . Dental phenotype in Jalili syndrome due to A C.1312 Dupc homozygous mutation in the CNNM4 gene. PloS One 2013; 8 (10): e78529.
Miller SA, Dykes DD, Polesky HF . A simple salting out procedure for extracting DNA From human nucleated cells. Nucleic Acids Res 1988; 16 (3): 1215.
Chang YF, Imam JS, Wilkinson MF . The nonsense-mediated Decay RNA surveillance pathway. Ann Rev Biochem 2007; 76: 51–74.
Coppieters F, Van Schil K, Bauwens M, Verdin H, De Jaegher A, Syx D et al. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med 2014; 16 (9): 671–680.
Huang L, Xiao X, Li S, Jia X, Wang P, Guo X et al. CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun 2012; 426 (4): 498–503.
Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet 2016; 53 (2): 98–110.
Lopez Torres L, Schorderet D, Valmaggia C, Todorova M . A Novel mutation in CNNM4 (G492C) associated with Jalili syndrome. Acta Ophthalmologica 2015; 93 (S255). Available at http://onlinelibrary.wiley.com/doi/10.1111/j.1755-3768.2015.0606/abstract.
Acknowledgements
We would like to thank our patients and their families for their participation. This study was funded by Shahid Beheshti University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Rahimi-Aliabadi, S., Daftarian, N., Ahmadieh, H. et al. A novel mutation and variable phenotypic expression in a large consanguineous pedigree with Jalili syndrome. Eye 30, 1424–1432 (2016). https://doi.org/10.1038/eye.2016.137
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2016.137
This article is cited by
-
Identification of a mutation in CNNM4 by whole exome sequencing in an Amish family and functional link between CNNM4 and IQCB1
Molecular Genetics and Genomics (2018)
