
Abstract
Purpose
X-linked juvenile retinoschisis (XLRS), a leading cause of juvenile macular degeneration, is characterized by a spoke-wheel pattern in the macular region of the retina and splitting of the neurosensory retina. This study aimed to identify the underlying genetic defect in a Chinese family with XLRS.
Methods
The proband underwent complete ophthalmic examinations, including fundus examination, fundus autofluorescence, and optical coherence tomography. DNA extracted from proband and his younger brother was screened for mutations in RS1 gene. The detected RS1 mutation was tested in all available family members and 200 healthy controls.
Results
Reduced visual acuity, spoke-wheel pattern at the fovea, and split retina were observed in the proband. A novel frameshift mutation c.206-207delTG in the RS1 gene, leading to a truncated protein (p.L69fs16X), was identified in the proband and his younger brother. This mutation was not found in any unaffected member or in the healthy controls. The mother of the proband was hemizygous for this mutant allele.
Conclusions
We identified a novel causative mutation of RS1 in a Chinese family with XLRS. This finding expands the mutation spectrum of RS1 and provides evidence for a phenotype–genotype study in XLRS.
Similar content being viewed by others
Introduction
X-linked juvenile retinoschisis (XLRS; OMIM 312700) is a rare macular dystrophy first described in 1898 by German ophthalmologist Josef Haas. It is the leading cause of juvenile macular degeneration and caused by mutations in the RS1 gene in Xp22.2 identified by Sauer in 1997 using positional cloning.1 The human RS1 gene contains six separate exons that encode a 224-amino-acid cell-surface protein known as retinoschisin (RS1).2, 3 RS1 is highly expressed by the retinal photoreceptor and bipolar cells and interacts with the surfaces of these cells to stabilize the organization of the retina.4, 5, 6
Pathogenic mutations of the RS1 gene lead to vitreoretinal dystrophies and progressive deterioration of vision. The disorder, which affects only males, has a prevalence of 1 in 5000–25 000 men.7 Female carriers rarely show vision impairment.8 Affected males show a spoke-wheel pattern in the fovea caused by cystic changes in the early stage (the first and second decades of life), leading to reduced visual acuity.9, 10 In later stages of the disease, patients display nonspecific macular atrophy because the coalescing cysts form a large central cavity. Approximately 50 % of cases show bilateral peripheral retinal lesions.9, 10 The complications include vitreous hemorrhage, choroidal sclerosis, retinal detachment, and neovascular glaucoma.11 The full-field electroretinogram (ERG) in patients with XLRS exhibits a relative preservation of an a-wave amplitude, which characterizes photoreceptor function, but a substantial reduction of the dark-adapted b-wave amplitude originating in inner retinal cell activity, which suggests that the main functional defect may occur after phototransduction, or does is inner retinal.12, 13 Because a XLRS individual with an identified RS1 mutation may have normal ERG, the diagnosis of XLRS cannot be based on ERG alone.14 Retinal fundus examination, optical coherence tomography (OCT), and fundus fluorescein angiography (FFA) are increasingly used in the diagnosis of hereditary retinal diseases.15 Coalescence of foveal schisis can be seen by fundus images and OCT scans in older affected patients.16, 17 Severely affected and older individuals display atrophic macular in the retinal pigment epithelium by taking FFA.18
In this study, we report on a Chinese family with XLRS, and based on clinical studies and molecular diagnostics, we identified a novel mutation c.206-207delTG in the RS1 gene, which caused a truncated protein (p.L69fs16X) and led to XLRS. This novel mutation expands the mutational spectrum, and the phenotype of the affected member of this family provides evidence for phenotype–genotype studies of XLRS.
Materials and methods
This study included two clinically affected individuals with retinoschisis, five unaffected relatives from a Chinese family (Figure 1), and 200 unrelated normal controls. All of the participants came from Hunan Jiahui Genetic Hospital. This study complied fully with the Tenets of the Declaration of Helsinki, and was approved by the Ethics Board of the State Key Laboratory of Medical Genetics of China. Informed consent was obtained from all participants.

Pedigree of X-linked juvenile retinoschisis Chinese family. Black symbols indicate affected individuals and white symbols indicate unaffected individuals. The arrow indicates the proband.
The proband, a 7-year-old boy, was a Chinese from Hunan province, in China. After a full pregnancy of the primigravid mother, he was born at a normal weight (approximately 3.5 kg), and normal height without hypoxia asphyxia. Consanguinity was excluded, and no chemical or maternal drug use and radiation or toxic exposure were noted during his mother’s pregnancy. He was 123 cm in height, 23 kg in weight, with normal intelligence, and normal physical development, but without neurobehavioral abnormalities or other malformation. He was referred to the eye clinic at 5 years old owing to the deterioration of his vision. His younger brother was 8 months old, and his eyes could not follow moving objects and suffered from anemia, but not Mediterranean anemia.
Clinical investigations
The proband underwent complete ophthalmic examinations, including fundus examination, fundus autofluorescence, and OCT.
Molecular genetics
Genomic DNA was isolated from peripheral blood samples, including all family members and controls, using standard extraction methods. The entire coding region and intron–exon boundaries of the RS1 gene (RefSeq NM_000330.3) were amplified by polymerase chain reaction (PCR). PCR products were directly sequenced with the ABI PRISM BigDye kit (Carlsbad, CA, USA) on an ABI3100/3130 DNA sequencer (Carlsbad, CA, USA). Sequences were analyzed using DNASTAR (Madison, WI, USA) software.
Results
Clinical findings
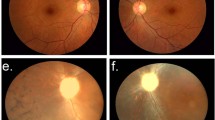
Eye examination showed that the proband had ametropia with Vos 0.08 and Vod 0.4; OD+1.0ODS|+1.00DC × 70, 0.4 2; OS+1.5ODS|+1.75DC × 70, 0.3. Fundus examination revealed that the binocular optic papilla was oval, boundary was clear, color was pink, C/D (cup/disc ratio) was 0.3 (reference φ 0.3), and A/V (retinal arteriovenous ratio) was 2/3 (reference=2/3). Fundus photography (Figure 2a) also showed a lack of macular reflex in both eyes. The right eye presented with pigment disorder and stellate cystic changes. Vitreous opacity, a hole in the inner layer and laser spot involved in the temporal retina could be seen in the left eye. Fundus photography with the RetCam showed temporal retinal detachment and several schisis at the 5 o’clock of the left eye. FFA (Figure 2b) revealed that the fluorescence in the posterior pole was low, and fluorescence showed leakage in the centers of both eyes. OCT (Figure 2c) revealed several cystic changes in the macular, and splitting of the neurosensory retina with several connected bridge-like retinal tissues that separated the retina into several regions of low reflex. After retinal detachment surgery, vision acuity was not improved.

Eye examination of the proband (III1). R: right eye; L: left eye. (a) Fundus photography, (b) fundus autofluorescence, and (c) optical coherence tomography.
The proband’s mother, a carrier, as well as all the unaffected family members, had normal visual acuity. The proband’s younger brother was too young for ophthalmic examination.
Molecular genetics
Direct sequencing of the PCR products of the proband showed a TG deletion at c.206-207 of the RS1 gene (Figure 3b), which was also displayed in his younger brother. This novel nucleotide change is predicted to create a frameshift at codon 69 with substitution of a leucine for a arginine and introduction of a putative stop codon-16 amino acids downstream in the translated protein (p.Arg69LeufsX16) (Figure 3d), and lead to the truncation of the last 137 amino acids (62% of the polypeptide). The proband’s mother was heterozygous for this mutation (Figure 3c) and had normal visual acuity. Neither other unaffected relatives nor the 200 healthy controls exhibited the mutation (Figure 3a).

Genetic characterization of the RS family. (a) Sequencing result of the normal individuals. (b) Sequencing result of the proband and his younger brother showing the two-base deletion (c.206-207delTG) in RS1. (c) Sequencing result of the proband’s mother showing heterozygous deletion (c.206-207delTG) in RS1. (d) Relative position of the mutation in RS1. (e) Evolutionary conservation of amino acids: the box shows loss of the conserved leucine residue and truncation of protein in the proband and in his younger brother.
Discussion
The RS1 protein contains several domains: a hydrophobic signal sequence encoded by exons 1 and 2 (amino acids 1–23), an RS1 domain encoded by exon 3 (amino acids 23–62), and a highly conserved discoidin domain encoded by exons 4–6 (amino acids 63–219).19, 20 In addition, a 5-amino-acid segment is located in the downstream of the discoidin domain.20, 21 The mature retinoschisin structure, a mature protein of 201 amino-acid residues (23 kDa), is derived from cleavage of the N-terminus of the 23-amino-acid hydrophobic leader sequence.19 The conserved discoidin domain is present in extracellular or transmembrane proteins, and is essential for stabilizing the retinal cell structure and establishing proper synaptic connectivity.22 It is implicated in cell adhesion and cell–cell interactions on membrane surfaces, such as the coagulation factors V and VIII, milk fat globules, and neuropilin and neurexins, and is also implicated in phospholipid binding.2, 10 The mutant protein encoded by mutant RS1 gene cannot fold properly and is probably accumulated both intracellularly and extracellularly, eventually leading to cystic changes and schisis formation.9, 23
To date, about 206 different mutations in the RS1 gene have been reported in patients with XLRS (HGMD Professional 2013.2). Most are missense mutations, and most of these missense mutations have been identified in the RS1 discoidin domain. Several structural models have been developed, and functional analysis indicates that mutations in the conserved discoidin domain result in a improperly folded, nonfunctional protein.2, 19 There are also nonsense mutations, insertions, deletions, and splice site mutations in the RS1 gene. The RS1 gene knockout mouse is consistent with human XLRS in showing schisis cavities and ERG decline, and shows structural and functional rescue at 14 months after injecting with AAV-RS1h at 14 days.24
In the present study, a deletion mutation (c.206-207delTG) in the RS1 gene was identified. This mutation cosegregated with the disease phenotype and was found neither in the healthy members of the family except the proband’s mother nor in the 200 healthy control individuals, indicating that the c.206-207delTG of RS1 is the pathogenic mutation of XLRS in this family. Truncation of retinoschisin protein associated with frameshift mutations caused by deletions has been frequently reported in XLRS individuals, such as c.194delA, c.195delT, c.219delA, c.300delG, c.371-374delAGAT, c.392-393delAA, c.416delA, and c.488delG.18, 25, 26, 27 These deletion mutations are all predicted to cause frameshift, thereby generating premature RS protein. Likewise, the two-base deletion mutation (c.206-207delTG) identified in this study occurs in the highly conserved discoidin domain leads to a premature stop at codon 84, and is expected to yield a truncated protein. The shortened polypeptide may fold improperly, leading to an nonfunctional product.25 Even extreme C-terminal nucleotide deletions, such as 655delT and 655-679del, could destroy the Cys219 residue of the discoidin domain.27 Progressive deterioration of vision, a spoke-wheel pattern in the fovea, and macular atrophy were found in all these male patients.
The proband exhibited the characteristic phenotype of XLRS—stellate cystic changes of the region of the retina, a splitting of the neurosensory retina, and visual deterioration. Most of genotype–phenotype studies about XLRS focused on b/a-ratio amplitudes, but not all individuals with an identified RS1 mutation had abnormal ERG.28, 29, 30 Although the genotype–phenotype relationship in XLRS is still controversial, many studies indicated that some mutations were more severe mutations, including which would cause major structural change, eliminate the production of retinoschisin protein or alter cysteine residue.27, 29, 30 The proband in our study showed several peripheral retinal schisis in the left eye, which was only exhibited in about 50 % of cases. Temporal retinal detachment was also found, and his visual acuity had gradually decreased since 5 years old. All these symptoms of the proband indicated that his phenotype was serious and this might be the result of the truncation protein encoded by c.206-207delTG in the RS1 gene. His mother was a carrier, but she had no ophthalmologic problems. Our results agree with those of previous studies in concluding that retinoschisis is an X-linked disease with recessive inheritance. It is unclear whether the anemia of the proband’s younger brother was associated with the disease.
In conclusion, we have identified a novel frameshift mutation c.206-207delTG in the RS1 gene. Our findings provide new information regarding the XLRS pathogenic mutation spectrum and evidence for a phenotype–genotype study in XLRS. The findings will also support clinical diagnosis and genetic counseling.

Change history
13 November 2014
This article has been corrected since Advance Online Publication and an erratum is also printed in this issue
References
Sauer CG, Gehrig A, Warneke-Wittstock R, Marquardt A, Ewing CC, Gibson A et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997; 17: 164–170.
Sergeev YV, Caruso RC, Meltzer MR, Smaoui N, MacDonald IM, Sieving PA . Molecular modeling of retinoschisin with functional analysis of pathogenic mutations from human X-linked retinoschisis. Hum Mol Genet. 2010; 19 (7): 1302–1313.
Vijayasarathy C, Sui R, Zeng Y, Yang G, Xu F, Caruso RC et al. Molecular mechanisms leading lo null-protein product from retinoschisin(RS1) singal-sequence mutants in X-linked retinoschisis (XlRS) disease. Hum Mutat 2010; 31 (11): 1251–1260.
Reid SN, Akhmedov NB, Piriev NI, Kozak CA, Danciger M, Farber DB . The mouse X-linked juvenile retinoschisis cDNA: expression in photoreceptors. Gene 1999; 227 (2): 257–266.
Khan NW, Jamison JA, Kemp JA, Sieving PA . Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001; 41 (28): 3931–3942.
Molday LL, Hicks D, Sauer CG, Weber BH, Molday RS . Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci 2001; 42 (3): 816–825.
Tsang SH, Vaclavik V, Bird AC, Robson AG, Holder GE . Novel phenotypic and genotypic findings in X-linked retinoschisis. Arch Ophthalmol 2007; 125 (2): 259–267.
George ND, Yates JR, Moore AT . Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996; 114 (3): 274–280.
Lesch B, Szabó V, Kánya M, Varsányi B, Somfai GM, Hargitai J et al. Truncation of retinoschisin protein associated with a novel splice site mutation in the RS1 gene. Mol Vis 2008; 14: 1549–1558.
Xu J, Gu H, Ma K, Liu X, Snellingen T, Sun E et al. R213W mutation in the retinoschisis 1 gene causes X-linked juvenile retinoschisis in a large Chinese family. Mol Vis 2010; 16: 1593–1600.
Grayson C, Reid SN, Ellis JA, Rutherford A, Sowden JC, Yates JR et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri-Rb1 cells. Hum Mol Genet 2000; 9 (12): 1873–1879.
Tantri A, Vrabec TR, Cu-Unjieng A, Frost A, Annesley WH Jr, Donoso LA . X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol 2004; 49 (2): 214–230.
Ziccardi L, Vijayasarathy C, Bush RA, Sieving PA . Loss of retinoschisin (RS1) cell surface protein in maturing mouse rod photoreceptors elevates the luminance threshold for light-driven translocation of transducin but not arrestin. J Neurosci 2012; 32 (38): 13010–13021.
Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT et al. X-Linked Juvenile Retinoschisis. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle 1993-2014.
Kortüm K, Kernt M, Reznicek L . Significance of ophthalmological imaging in common hereditary retinal diseases. Klin Monbl Augenheilkd 2013; 230 (3): 223–231.
Apushkin MA, Fishman GA, Janowicz MJ . Correlation of optical coherence tomography findings with visual acuity and macular lesions in patients with X-linked retinoschisis. Ophthalmology 2005; 112 (3): 495–501.
Pierro L, Fogliato G, Gagliardi M, Codenotti M . Correspondence to: Use of spectral-domain optical coherence tomography to differentiate acquired retinoschisis from retinal detachment in difficult cases. Retina 2013; 33 (6): 1290–1291.
Li X, Ma X, Tao Y . Clinical features of X-linked juvenile retinoschisis in Chinese families associated with novel mutations in the RS1 gene. Mol Vis 2007; 13: 804–812.
Wu WW, Wong JP, Kast J, Molday RS . RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem 2005; 280 (11): 10721–10730.
Wu WW, Molday RS . Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X-linked retinoschisis. J Biol Chem 2003; 278 (30): 28139–28146.
Skorczyk A, Krawczyński MR . Four novel RS1 gene mutations in Polish patients with X-linked juvenile retinoschisis. Mol Vis 2012; 18: 3004–3012.
Takada Y, Vijayasarathy C, Zeng Y, Kjellstrom S, Bush RA, Sieving PA . Synaptic pathology in retinoschisis knockout (Rs1-/y) mouse retina and modification by rAAV-Rs1 gene delivery. Invest Ophthalmol Vis Sci 2008; 49 (8): 3677–3686.
Wang T, Zhou A, Waters CT, O'Connor E, Read RJ, Trump D . Molecular pathology of X linked retinoschisis: mutations interfere with retinoschisin secretion and oligomerisation. Br J Ophthalmol 2006; 90 (1): 81–86.
Kjellstrom S, Bush RA, Zeng Y, Takada Y, Sieving PA . Retinoschisin gene therapy and natural history in the Rs1h-KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci 2007; 48 (8): 3837–3845.
Hiriyanna KT, Bingham EL, Yashar BM, Ayyagari R, Fishman G, Small KW et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change. Hum Mutat 1999; 14 (5): 423–427.
Inoue Y, Yamamoto S, Okada M, Tsujikawa M, Inoue T, Okada AA et al. X-linked retinoschisis with point mutations in the XLRS1 gene. Arch Ophthalmol 2000; 118 (1): 93–96.
The Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum Mol Genet 1998; 7 (7): 1185–1192.
Vincent A, Robson AG, Neveu MM, Wright GA, Moore AT, Webster AR et al. A phenotype-genotype correlation study of X-linked retinoschisis. Ophthalmology 2013; 120 (7): 1454–1464.
Bowles K, Cukras C, Turriff A, Sergeev Y, Vitale S, Bush RA et al. X-linked retinoschisis: RS1 mutation severity and age affect the ERG phenotype in a cohort of 68 affected male subjects. Invest Ophthalmol Vis Sci 2011; 52 (12): 9250–9256.
Sergeev YV, Vitale S, Sieving PA, Vincent A, Robson AG, Moore AT et al. Molecular modeling indicates distinct classes of missense variants with mild and severe XLRS phenotypes. Hum Mol Genet 2013; 22 (23): 4756–4767.
Acknowledgements
We thank the patients’ families and healthy controls for their participation in our study. We also thank Ranhui Duan for manuscript revising. This study was supported by the National Natural Science Foundation of China (81270706) and (81271944).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Huang, Y., Mei, L., Gui, B. et al. A novel deletion mutation in RS1 gene caused X-linked juvenile retinoschisis in a Chinese family. Eye 28, 1364–1369 (2014). https://doi.org/10.1038/eye.2014.196
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2014.196
