
Abstract
Purpose
To describe the clinical and imaging presentation, pitfalls in the diagnosis of choroidal extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT), as well as the therapeutic management and prognosis.
Methods
A retrospective case review of nine choroidal MALT lymphomas was performed. Initial clinical presentation and imaging findings of these histologically confirmed cases of lymphoma were analyzed. Treatment methods, time to diagnosis, systemic work-up, and treatment prognosis were assessed.
Results
Initial presentation was essentially blurred vision. The features described on examination were: anterior and posterior scleritis, iridocyclitis, choroidal infiltration, and exudative retinal detachment. Fluorescein and indocyanine green angiography as well as ultrasonography and optic coherence tomography provided arguments in favor of the diagnosis. Biopsy sites included conjunctiva, Tenon’s capsule, deep scleral tissue, episclera, lacrimal gland, and choroid. Treatment mostly consisted of a combination of chemotherapy and radiotherapy. The mean time to diagnosis was 12 months.
Conclusions
Owing to the insidious onset of these tumors and their ability to simulate other conditions, the diagnosis is commonly delayed. The prognosis is generally good and treatment is effective in the case of localized lymphoma.
Similar content being viewed by others
Introduction
Non-Hodgkin lymphoma (NHL) is a general term for a large group of malignancies of lymphoid tissue. Although it usually affects the lymphatic system, extranodal sites are common and may involve almost all tissues. Although lymphoma of the ocular region is a relatively rare condition, NHL is the most common type and typically involves mucosa-associated lymphoid tissue (MALT). MALT lymphoma usually involves the conjunctiva, lacrimal gland, and orbit, but intraocular choroidal involvement is rare and has rarely been reported. As the initial presentation is frequently nonspecific and insidious,1, 2, 3 this form of MALT lymphoma is particularly difficult to diagnose and treat. Based on a better knowledge of this condition, clinicians may be able to recognize this disease at an early stage, at which treatment may prevent systemic dissemination of the disease or transformation into a more aggressive lymphoma.4, 5, 6 In this study, we report the clinical findings, medical imaging, and therapeutic management of nine patients with a histologically confirmed diagnosis of choroidal MALT lymphoma.
Patients and methods
This study is a retrospective case review of nine consecutive patients with choroidal MALT lymphoma. All patients were seen in a referral center between 1998 and 2010. Patients’ medical records were reviewed for the following inclusion criteria: histologically confirmed diagnosis of MALT lymphoma, primary choroidal involvement. Evidence of MALT lymphoma was based on a set of arguments such as morphology, immunohistochemistry, and molecular biology. Choroidal involvement was documented clinically and on ultrasonography and fluorescein angiography. All patients were referred to our center after failure of first-line medical management. The following data were collected for each patient: age, gender, general comorbidity, presenting clinical features, initial treatment method and investigations, visual acuity testing, slit-lamp and fundus examination, medical or surgical interventions, and locoregional and systemic staging assessment. Pathological examination included biopsy, grading, and classification of the lymphoma, conventional histology, immunohistochemistry, and PCR for immunoglobulin light chain gene rearrangement. The time to diagnosis was defined as the time elapsed between onset of symptoms and histological diagnosis of marginal zone lymphoma. At final follow-up, remission status, relapse, and survival were assessed. Disease was considered resolved when patients did not present any recurrence of initial features or any other lymphoma site (or systemic spread) at last follow-up, corresponding to criteria of complete remission.
Results
Nine patients (11 eyes) with choroidal MALT lymphoma were identified. Mean age at presentation was 56 years (median, 54 years; range: 40–73 years). Mean follow-up was 74.2 months (median 60 months; range: 6–156 months). Follow-up was longer than 5 years for five patients. Two patients had bilateral eye involvement. Mean initial visual acuity was 20/50. Mean final acuity was 20/50. Nine patients presented progressive painless visual impairment and one patient reported isolated retrobulbar headache. Other symptoms included metamorphopsia and floaters. Clinical findings are summarized in Table 1. Examination of the anterior segment revealed chemosis in two patients and iridocyclitis with pseudohypopyon in two other patients. One eye had features of anterior scleritis. In one patient, the initial presentation also included red eye and epiphora. All nine patients had choroidal infiltrates on fundus examination (Figure 1). The other features on initial examination were: exudative retinal detachment (three cases), choroidal thickening (three cases), macular choroidal neovascularization (one case), optic disc swelling (two cases), and venous stasis retinopathy (one case).

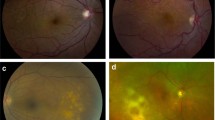
Fundus photography of bilateral choroidal involvement of choroidal MALT lymphoma. In the left eye (upper left): posterior choroidal infiltration. In right the eye (upper right): exsudative retinal detachment with peripheral cream-colored choroidal infiltrates. On the bottom left, green-light fundus photography of choroidal infiltrates; on the bottom right, same eye fluorescein angiography (1 : 02) showing the same features.
All patients underwent fluorescein angiography, which confirmed fundus examination findings in every case (Figures 1 and 2). On fluorescein angiography, patients typically had localized or diffuse thickening of the choroid with heterogeneous hyperfluorescence and alterations or migrations of retinal pigment epithelium (tiger-like pattern). These choroidal lesions remained hypofluorescent on ICG angiography. The eye with the venous stasis retinopathy was confused with extrinsic compression. B-scan ultrasonography was performed in all cases and showed diffuse, irregular scleral thickening, isoechoic to normal choroid, possibly associated with swelling of Tenon’s capsule and conjunctiva (2/10) (Figure 2). These features were confirmed by orbital MRI, showing a hyperintense area of thickened choroid with mild gadolinium enhancement. Detailed angiographic findings are summarized in Table 2. Two patients underwent extensive laboratory and imaging investigations (that gave negative results) for suspicion of acute uveitis. They were subsequently treated with topical and systemic corticosteroids, with only temporary improvement of visual acuity. Complementary investigations in one patient in this series with metamorphopsia and vision loss showed features of unilateral macular choroidal neovascularization (Figure 3). The initial diagnosis was that of age-related macular degeneration and the patient underwent repeated intravitreal anti-VEGF therapy.

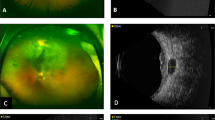
Upper left: fluorescein angiography (0 : 18). In this case, choroidal infiltration is more diffuse, and may be harder to identify; on the upper right, later time (6 : 06) of the same eye. On the bottom: B-Scan ultrasonography of a choroidal MALT lymphoma. Hypoechogenic and heterogenous parietal thickening, with no scleral breach.

Fluorescein angiography sequence showing unilateral macular choroidal neovascularization. Initial diagnosis was age-related macular degeneration.
Local and regional staging assessment revealed conjunctival involvement in three cases and orbital involvement in two cases. Lacrimal gland infiltration was reported in two patients. The systemic work-up included chest, abdomen and pelvis CT, bone marrow aspirate, bone marrow biopsy (all patients), bone scintigraphy (three patients), PET scan (six patients), gastrointestinal endoscopy and colonoscopy (seven patients). Systemic work-up was negative in all cases. All lymphomas were histologically confirmed. Biopsy sites included conjunctiva (five cases), Tenon’s capsule (three cases), deep scleral tissue (three cases), episclera (two cases), lacrimal gland (one case), ciliary body (one case), and choroid (one case). In one case, the first biopsy was negative and the diagnosis was subsequently confirmed by repeat biopsy from the same site. All patient samples demonstrated low grade B-cell type malignant lymphoma, CD20+++, CD5+. Pathological features are described in Table 2. Treatment included external radiotherapy in three patients, exclusive chemotherapy in three cases, and a combination of radiotherapy and systemic chemotherapy in three patients. Chemotherapy protocols used various combinations of drugs, mostly associated with rituximab. These protocols are reported in Table 3. Patients were treated with 30 Gy of irradiation in 1.5 Gy fractions over 4 weeks.
Persistent complete remission was achieved in all patients at the end of follow-up. Only one patient experienced reactivation of choroidal neovascularization that was subsequently controlled by repeated anti-VEGF therapy. The mean time to diagnosis was 12 months (median 12 months; (range: 2–24 months)). Overall visual outcome results are presented in Table 2.
Discussion
As choroidal MALT lymphoma is a very rare disease, only a small number of cases have been reported in the literature.1, 2, 3, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 This series of nine consecutive patients highlights the very heterogeneous initial clinical features. All patients in this study initially had very common and nonspecific symptoms, such as progressive blurred vision, floaters, red eye, or epiphora. All patients also reported a fairly insidious onset, often resulting in delayed medical consultation. These nonspecific symptoms can correspond to a large range of diseases, making it difficult for the clinician to immediately suspect a neoplastic disease.
In contrast, some patients presented with particular clinical features, highly suggestive of other specific diseases, leading to inappropriate work-up and therapeutic management. For example, a 73-year-old female patient presented with unilateral metamorphopsia, leading to investigation of possible age-related macular degeneration. Fluorescein angiography and optic coherence tomography (OCT) showed typical macular choroidal neovascularization, in favor of the initial, incorrect diagnosis. This patient then received intravitreal anti-VEGF therapy and the diagnosis was reviewed only after three injections, in view of the only temporary and limited benefit of treatment. A combination of scleritis and uveitis appears to be a common, misleading presentation. Some authors1, 8, 20 have described a masquerade syndrome, associated with MALT-type lymphoma, comprising features of posterior uveitis associated with chorioretinal infiltration, anterior scleritis associated with uveal effusion syndrome, and bilateral panuveitis with serous retinal detachment. Imaging features, including orbital MRI and ultrasonography, may also be confusing as they can depict irregular scleral thickening, compatible with posterior scleritis. Two cases in the present series presented non-resolving uveitis: the first patient presented inflammation of the vitreous body associated with choroidal infiltrates, in favor of white dots syndrome. Extensive assessment, including diagnostic vitrectomy, was initially negative and systemic corticosteroid therapy was unsuccessful. Examination of the second case also showed choroidal infiltration simulating chorioretinitis tuberculosis, subsequently treated by 6-months of anti-tuberculosis therapy. The subsequent onset of exudative retinal detachment raised the suspicion of lymphoma. Another study reports a case of choroidal lymphoma masquerading as anterior ischemic optic neuropathy,12 presenting optic nerve swelling on examination. Similarly, our series included two cases of optic nerve edema, one of which simulated venous stasis retinopathy, leading to investigation of possible extrinsic compression. Despite the rarity of this tumor, choroidal involvement of MALT lymphoma should be included in the differential diagnosis, particularly in patients with a non-resolving or recurrent ocular disease.1, 2, 3, 8, 10, 12, 14
The imaging work-up in most cases of this series included fluorescein and indocyanine green angiography. Fluorescein angiography mainly revealed multiple cream-colored choroidal infiltrates, macular edema, serous retinal detachment, diffuse infiltration with choroidal tiger-like pattern, and thickening of the choroid with choroidal folds, which, in one patient, led to an incorrect diagnosis of extrinsic compression. Indocyanine green angiography showed choroidal lesions and early geographic hypofluorescent areas with pin points. Some authors have reported that indocyanine green angiography is much more reliable than fluorescein angiography to visualize choroidal involvement and would be superior to the diagnosis and follow-up of patients with choroidal lymphoma.17 In most cases of the present series, both techniques showed good sensitivity to identify the suspected condition. In two patients, however, early fluorescein and indocyanine green angiographies were considered to be normal, while subsequent investigations revealed uveal effusion syndrome with significant choroidal thickening. None of these features were truly specific, but, together with the clinical findings, they provided a strong set of arguments in favor of choroidal lymphoma. Macular involvement, such as serous retinal detachment or choroidal neovascularization, were easily confirmed on OCT,16 whereas scleral and episcleral alterations were well documented on ultrasonography and MRI.
Features of locoregional extension may also provide arguments suggestive of MALT lymphoma. Six patients in this series presented conjunctival and orbital extension with typical features of lymphoma, guiding the subsequent investigations. Histological evidence remains mandatory for the diagnosis of choroidal MALT lymphoma. The first option consists of performing superficial biopsy on the most accessible affected site. Conjunctival or Tenon’s capsule biopsy is a safe and minimally invasive technique, and is frequently considered as first-line procedure.1, 8, 9, 10 These superficial biopsies were mostly conclusive. Superficial and deep episcleral or scleral biopsies were also performed in several patients in this series, and may constitute an alternative when no superficial site is available. Orbital and lacrimal gland biopsy, although more invasive, may also be useful. In patients with an inconclusive conjunctival biopsy, a second biopsy must be performed before undertaking more invasive surgery. In one patient in this series, histological confirmation of the diagnosis was only obtained after two biopsies at the same site. When no superficial site is available, more invasive direct biopsy should be considered. Endoretinal and transretinal choroidal biopsy for the management of atypical presentation of uveitis5, 20, 21, 22, 23 has been described by several authors, who reported that choroidal biopsy could be very helpful and was associated with a low risk of complications. Transretinal choroidal biopsy and diagnostic vitrectomy were performed in one patient in this series following inconclusive first-line conjunctival biopsy. Vitrectomy was negative but the tissue specimen provided an accurate diagnosis of MALT lymphoma. Pathological examination included conventional histology, immunochemistry and PCR techniques that confirmed the diagnosis of marginal zone B-cell lymphoma in every case.24
Apart from almost systematic locoregional extension, the staging work-up did not reveal any evidence of systemic lymphoma. Therapeutic management was mostly based on combination chemotherapy and local radiotherapy, with good antitumor efficacy, as all patients achieved complete remission at last follow-up. Although conventional external radiotherapy remains the gold standard treatment, exclusive chemotherapy based on anti-CD20 regimen is increasingly used. In 2009, Paik et al25 showed that chemotherapy alone was as effective as radiotherapy for adnexal MALT lymphoma, with fewer ocular side effects. In terms of visual prognosis, several patients obtained rapid visual recovery, while those patients with more critical complications such as massive retinal detachment were not improved by treatment. None of the patients in this series required more radical surgery, such as enucleation.
In conclusion, choroidal involvement of MALT lymphoma may present various and nonspecific features. Owing to its insidious onset and its ability to simulate other conditions, the diagnosis is commonly delayed. Clinicians must be aware of the possibility of a masquerade syndrome of ocular lymphoma in a patient presenting non-resolving or atypical symptoms. A better knowledge of suggestive imaging findings would also facilitate early diagnosis. Overall, persistent signs and symptoms of scleritis or uveitis not responding to standard treatment should lead to prompt biopsy. All accessible sites should be biopsied to avoid more invasive and risky choroidal biopsy. Referral centers may provide significant assistance at this stage. The low relapse rate and rapid functional recovery demonstrate the need for early diagnosis and specific treatment to ensure a good prognosis and prevent systemic spread.

References
Rasić DM, Stanković Z, Terzić T, Kovacević D, Koturović Z, Marković V . Primary extranodal marginal zone lymphoma of the uvea associated with massive diffuse epibulbar extension and focal infiltration of the optic nerve and meninges, clinically presented as uveitis masquerade syndrome: a case report. Med Oncol 2010; 27 (3): 1010–1016.
Kase S, Saito W, Saito A, Ohno S . Uveal effusion syndrome caused by choroidal invasion of malignant lymphoma. Jpn J Ophthalmol 2010; 54 (1): 109–110.
Hammoudi DS, Tucker NA, Fernandes B, McGowan H . Orbital lymphoma presenting with choroidal detachments. Ophthal Plast Reconstr Surg 2010; 26 (6): 479–481.
Deutsch AJ, Steinbauer E, Hofmann NA, Strunk D, Gerlza T, Beham-Schmid C et al. Chemokine receptors in gastric MALT lymphoma: loss of CXCR4 and upregulation of CXCR7 is associated with progression to diffuse large B-cell lymphoma. Mod Pathol 2012; 26 (2): 182–194.
Esmaeli B, McLaughlin P, Pro B, Samaniego F, Gayed I, Hagemeister F et al. Prospective trial of targeted radioimmunotherapy with Y-90 ibritumomab tiuxetan (Zevalin) for front-line treatment of early-stage extranodal indolent ocular adnexal lymphoma. Ann Oncol 2009; 20 (4): 709–714.
Zucca E, Bertoni F, Roggero E, Cavalli F . The gastric marginal zone B-cell lymphoma of MALT type. Blood 2000; 96 (2): 410–419.
García-Alvarez C, Saornil MA, Blanco G, Méndez MC, López-Lara F . Extranodal B-cell uveal lymphoma with extraocular involvement. Can J Ophthalmol 2009; 44 (2): 213–214.
Gaucher D, Bodaghi B, Charlotte F, Schneider C, Cassoux N, Lemaitre C et al. MALT-type B-cell lymphoma masquerading as scleritis or posterior uveitis. J Fr Ophtalmol 2005; 28 (1): 31–38.
Charlotte F, Doghmi K, Cassoux N, Ye H, Du MQ, Kujas M et al. Ocular adnexal marginal zone B cell lymphoma: a clinical and pathologic study of 23 cases. Virchows Arch 2006; 448 (4): 506–516.
Ramulu P, Iliff NT, Green WR, Kuo IC . Asymptomatic conjunctival mucosa-associated lymphoid tissue-type lymphoma with presumed intraocular involvement. Cornea 2007; 26 (4): 484–486.
Lee HB, Pulido JS, Buettner H, Salomão D, Zent CS, Link TP . Intravascular B-cell lymphoma (angiotropic lymphoma) with choroidal involvement. Arch Ophthalmol 2006; 124 (9): 1357–1359.
Tavallali A, Shields CL, Bianciotto C, Shields JA . Choroidal lymphoma masquerading as anterior ischemic optic neuropathy. Eur J Ophthalmol 2010; 20 (5): 959–962.
Mudhar HS, Sethuraman C, Khan MD, Intraocular JanSU . Pan-uveal intravascular large B-cell lymphoma associated with choroidal infarction and choroidal tri-lineage extramedullary haemtopoiesis. Histopathology 2007; 51 (2): 275–279.
Apte RS, Al-Abdulla NA, Green WR, Schachat AP, DeJong MR, DiBernardo C et al. Systemic non-Hodgkin B-cell lymphoma encountered as a vanishing choroidal mass. Arch Ophthalmol 2005; 123 (1): 105–109.
Sarraf D, Jain A, Dubovy S, Kreiger A, Fong D, Paschal J . Mucosa-associated lymphoid tissue lymphoma with intraocular involvement. Retina 2005; 25 (1): 94–98.
Williams BK Jr, Tsui I, McCannel TA . Spectral-domain optical coherence tomography of conjunctival mucosa-associated lymphoid tissue lymphoma with presumed choroidal involvement. Graefes Arch Clin Exp Ophthalmol 2010; 248 (12): 1837–1840.
Saatci AO, Arikan G, Ozcan MA, Ozkal S, Kargi A, Undar B . Indocyanine green angiographic features of systemic non-Hodgkin's lymphoma and bilateral choroidal involvement. Ophthalmic Surg Lasers Imaging. 2006; 37 (3): 236–239.
Ozcan AA, Paydas S, Soylu M, Yavuz S . Bilateral choroidal infiltration from indolent non-Hodgkin's lymphoma: a rapid course with poor prognosis. Leuk Lymphoma 2005; 46 (4): 615–617.
Barbón García JJ, Viña Escalar C, Menéndez Fernández CL, Fernández Alvarez C, Carballo Fernández C, Villarreal Renedo PM . Uveal lymphoid infiltration with systemic extension. Arch Soc Esp Oftalmol 2003; 78 (3): 173–176.
Cassoux N, Charlotte F, Rao NA, Bodaghi, Merle-Beral H, LeHoang P . Endoretinal biopsy in establishing the diagnosis of uveitis: a clinicopathologic report of three cases. Ocul Immunol Inflamm 2005; 13 (1): 79–83.
Sen J, Groenewald C, Hiscott PS, Smith PA, Damato BE . Transretinal choroidal tumor biopsy with a 25-gauge vitrector. Ophthalmology 2006; 113 (6): 1028–1031.
Johnston RL, Tufail A, Lightman S, Luthert PJ, Pavesio CE, Cooling RJ et al. Retinal and choroidal biopsies are helpful in unclear uveitis of suspected infectious or malignant origin. Ophthalmology 2004; 111 (3): 522–528.
Lim LL, Suhler EB, Rosenbaum JT, Wilson DJ . The role of choroidal and retinal biopsies in the diagnosis and management of atypical presentations of uveitis. Trans Am Ophthalmol Soc 2005; 103: 84–91 discussion 91-2.
Coupland SE, Joussen A, Anastassiou G, Stein H . Diagnosis of a primary uveal extranodal marginal zone B-cell lymphoma by chorioretinal biopsy: case report. Graefes Arch Clin Exp Ophthalmol 2005; 243 (5): 482–486.
Paik JS, Cho WK, Lee SE, Choi BO, Jung SE, Park GS et al. Ophthalmologic outcomes after chemotherapy and/or radiotherapy in non-conjunctival ocular adnexal MALT lymphoma. Ann Hematol 2012; 91 (9): 1393–1401.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Loriaut, P., Charlotte, F., Bodaghi, B. et al. Choroidal and adnexal extranodal marginal zone B-cell lymphoma: presentation, imaging findings, and therapeutic management in a series of nine cases. Eye 27, 828–835 (2013). https://doi.org/10.1038/eye.2013.74
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2013.74
